


Содержание
Перейти к:
 Р. П. Мясников,
Н. Н. Кузина,
Д. А. Нефедова,
А. В. Киселева,
О. В. Куликова,
А. Н. Мешков,
М. М. Кудрявцева,
Е. А. Мершина,
М. Г. Дивашук,
Е. В. Рыжкова,
М. С. Харлап,
О. М. Драпкина
Р. П. Мясников,
Н. Н. Кузина,
Д. А. Нефедова,
А. В. Киселева,
О. В. Куликова,
А. Н. Мешков,
М. М. Кудрявцева,
Е. А. Мершина,
М. Г. Дивашук,
Е. В. Рыжкова,
М. С. Харлап,
О. М. Драпкина https://doi.org/10.15829/1560-4071-2023-5648
EDN: JSXHYA
Перейти к:
Наследственные кардиомиопатии (КМП) — это группа гетерогенных заболеваний, характеризующихся патологией сердечной мышцы, не обусловленной ишемической болезнью сердца, гипертензией, клапанными и врожденными пороками. Благодаря развитию методов визуализации и молекулярногенетической диагностики стало понятно, что для многих КМП характерен фенотипический и генотипический "перекрест". И хотя генетическая составляющая не всегда определяет конкретный фенотип заболевания, в настоящее время доказано, что генетическое тестирование играет существенную роль в стратификации риска, определении прогноза и тактики ведения пациентов, а также проведении семейного скрининга. Принимая во внимание высокую диагностическую и прогностическую значимость генотипа, в последние годы были предложены новые стратегии классификации КМП, основанные на генотипе, а не на фенотипе. Одним из примеров такого генотип-специфического подхода является выделение десмоплакиновой КМП как отдельной самостоятельной нозологической единицы. В статье представлен клинический случай семьи, в трех поколениях которой выявлен патогенный вариант гена DSP (p.Gln948LysfsTer29), приводящий к развитию специфического фенотипа КМП. Проведено комплексное обследование, продемонстрирована стадийность естественного течения заболевания, а также предложена тактика ведения пациентов с десмоплакиновой КМП.
Мясников Р.П., Кузина Н.Н., Нефедова Д.А., Киселева А.В., Куликова О.В., Мешков А.Н., Кудрявцева М.М., Мершина Е.А., Дивашук М.Г., Рыжкова Е.В., Харлап М.С., Драпкина О.М. Десмоплакин и особенности течения десмоплакиновой кардиомиопатии. Российский кардиологический журнал. 2023;28(11):5648. https://doi.org/10.15829/1560-4071-2023-5648. EDN: JSXHYA
Myasnikov R.P., Kuzina N.N., Nefedova D.A., Kiseleva A.V., Kulikova O.V., Meshkov A.N., Kudryavtseva M.M., Mershina E.A., Divashuk M.g., Ryzhkova E.V., Kharlap M.S., Drapkina O.M. Desmoplakin and features of desmoplakin cardiomyopathy. Russian Journal of Cardiology. 2023;28(11):5648. (In Russ.) https://doi.org/10.15829/1560-4071-2023-5648. EDN: JSXHYA
Наследственные кардиомиопатии (КМП) — это группа гетерогенных заболеваний, характеризующихся патологией сердечной мышцы с ее структурными и/или функциональными нарушениями, не обусловленными ишемической болезнью сердца, гипертензией, клапанными пороками и врожденными заболеваниями [1]. На сегодняшний день в клинической практике преимущественно используются классификации КМП, основанные на фенотипе, согласно которым выделяют дилатационную, гипертрофическую, рестриктивную, аритмогенную и другие редкие формы КМП. Тем не менее, несмотря на значительный прогресс в понимании этиологии и механизмов патогенеза КМП, достигнутый в последние десятилетия, основным недостатком современных классификаций КМП остается фенотипический и генотипический "перекрест" данных заболеваний [2]. Благодаря развитию методов визуализации (в первую очередь магнитно-резонансной томографии (МРТ) сердца с контрастным усилением) и молекулярно-генетической диагностики, стало понятно, что наследственные заболевания сердца с похожими клинико-морфологическими характеристиками являются генетически гетерогенными, а варианты в одних и тех же генах могут приводить к формированию различных фенотипов КМП. В то же время, хотя генетическая составляющая не всегда определяет конкретный фенотип заболевания, литературные данные свидетельствуют о том, что некоторые гены ассоциированы с более высоким риском развития таких осложнений, как прогрессирующая сердечная недостаточность (СН), жизнеугрожающие аритмии и внезапная сердечная смерть (ВСС) [3-6]. В связи с этим в настоящее время молекулярногенетическое исследование является важным этапом диагностики КМП, поскольку выявление патогенных/вероятно патогенных вариантов генов играет существенную роль в стратификации риска, определении прогноза и выборе индивидуального подхода к тактике ведения пациента, в частности, профилактике ВСС. Кроме этого, генетическое тестирование также позволяет проводить семейный скрининг с целью выявления пациентов на ранней бессимптомной и преморфологической стадии [7].
Принимая во внимание высокую диагностическую и прогностическую значимость генотипа, в последние годы появились новые стратегии классификации КМП, основанные на генотипе, а не на фенотипе [2]. Одним из примеров такого нового генотип-специфического подхода является выделение десмоплакиновой КМП как отдельной самостоятельной нозологической единицы [8][9].
Десмоплакин — белок, являющийся одним из основных структурных компонентов десмосом в кардиомиоцитах и эпидермисе, который кодируется геном DSP. Основная роль десмоплакина заключается в прикреплении промежуточных филаментов к десмосомным бляшкам. В сердечной мышце десмоплакин локализуется в десмосомах во вставочных дисках, которые механически соединяют кардиомиоциты в синцитий. Нарушения десмосомальных контактов в результате продукции дефектного десмоплакина снижают устойчивость кардиомиоцитов к механическим воздействиям, что ведет к разрыву синцития и гибели кардиомиоцитов с последующим замещением их фиброзной тканью [10].
Первым описанным в литературе заболеванием, ассоциированным с вариантами гена DSP, является синдром Карвахаля (Carvajal) — аутосомнорецессивный кожно-кардиальный синдром, характеризующийся дилатационной КМП (ДКМП), ладонно-подошвенной кератодермией и шерстистокурчавыми волосами [11]. В дальнейшем была выявлена взаимосвязь с развитием ДКМП, аритмогенной КМП (АКМП) и некомпактной КМП, причем пациенты с вариантами в гене DSP демонстрировали более злокачественный аритмогенный фенотип даже при отсутствии явной дисфункции и дилатации левого желудочка (ЛЖ) [12-14]. Кроме того, в серии клинических случаев у этих пациентов неоднократно были описаны эпизоды острого, рецидивирующего миокардиального повреждения ("hot phases"), которые вносили свой вклад в прогрессирование распространенных фиброзных изменений миокарда [15][16].
Принимая во внимание данные особенности, в последние годы был опубликован ряд работ, в которых авторами предложено выделять "десмоплакиновую КМП" как отдельную самостоятельную форму КМП, характеризующуюся специфическими клиническими и инструментальными признаками, такими как преимущественное поражение ЛЖ с обширным субэпикардиальным фиброзом, высокий риск развития жизнеугрожающих желудочковых аритмий и повторные эпизоды острого повреждения миокарда [8, 9]. Тем не менее многие вопросы, касающиеся, в частности, стратификации риска и тактики ведения этих пациентов, в настоящее время остаются нерешенными.
Ниже представлен клинический случай семьи, в трех поколениях которой выявлен патогенный вариант гена DSP, приводящий к развитию специфического фенотипа КМП. Нами было проведено комплексное клинико-инструментальное обследование и молекулярно-генетическое тестирование, продемонстрирована стадийность естественного течения болезни, а также предложена тактика ведения пациентов с десмоплакиновой КМП.
Исследование включало три поколения семьи с генетически детерминированной КМП и было одобрено независимым этическим комитетом ФГБУ "НМИЦ ТПМ" Минздрава России. Все участники подписали информированное согласие на участие в исследовании и обработку персональных данных. Всем участникам было проведено клиникоинструментальное обследование, включающее в себя электрокардиографическое исследование (ЭКГ), суточное мониторирование ЭКГ по Холтеру (ХМЭКГ), эхокардиографическое исследование (ЭхоКГ). Пробанду была выполнена МРТ сердца по стандартному протоколу с отсроченным внутривенным контрастированием и проведением Т1-, Т2-, ECVкартирования. Молекулярно-генетическое исследование проведено с помощью секвенирования следующего поколения на NextSeq 550 (Illumina, США) с использованием таргетной панели, включающей гены, ассоциированные с разными формами КМП [17]. Биоинформатический анализ и клиническая интерпретация описаны ранее [17]. Валидация выявленного варианта была проведена методом секвенирования по Сенгеру на Applied Biosystem 3500 Genetic Analyzer (Thermo Fisher Scientific, США).
Данные анамнеза, клинического и инструментального обследования
Пробанд — пациентка 43 лет, нормостенического телосложения. Рост 173 см, вес 65 кг. В возрасте 34 лет (2013г), после третьих родов, пациентку начали беспокоить приступообразные боли за грудиной, возникающие без четкой связи с провоцирующими факторами, однако по этому поводу к врачам не обращалась. С 2019г появилась одышка при физической нагрузке. В августе 2020г в связи с развитием интенсивного болевого синдрома за грудиной, сопровождающегося нарастанием одышки и учащенным аритмичным сердцебиением, пациентка была госпитализирована в стационар по месту жительства. На ЭКГ при поступлении — синусовый ритм с частотой сердечных сокращений (ЧСС) 65 уд./мин, частая желудочковая экстрасистолия (ЖЭС). Анализ на высокочувствительный тропонин двукратно отрицательный. При проведении ЭхоКГ была выявлена незначительная дилатация ЛЖ (конечно-диастолический размер 5,3 см, конечно-диастолический объем (КДО) 150 мл), диффузный гипокинез миокарда ЛЖ со снижением фракции выброса (ФВ) до 50%. При ХМЭКГ регистрировалась частая ЖЭС (3,5 тыс./сут.). По данным коронарографии коронарные артерии были интактны. После выписки пациентка наблюдалась кардиологом, проведена экспертная ЭхоКГ, при которой впервые описаны признаки некомпактного миокарда (критерий Jenni [18]). В октябре 2020г была выполнена МРТ сердца с гадолинием — выявлено снижение ФВ ЛЖ до 44%, дилатация полости ЛЖ (КДО 196 мл), повышенная трабекулярность миокарда в области боковой стенки ЛЖ, удовлетворяющая критериям некомпактности (критерий Petersen [19]), при отсроченном контрастировании определялась обширная циркулярная зона субэпикардиального контрастирования миокарда ЛЖ в базальных и средних сегментах. По результатам проведенных исследований был выставлен диагноз "некомпактная КМП", инициирована многокомпонентная терапия СН (валсартан/сакубитрил, эплеренон, метопролола сукцинат), с антиаритмической целью назначен пропафенон 75 мг 2 раза/сут. Также пациентка консультирована хирургом-аритмологом — показаний для имплантации кардиовертера-дефибриллятора (КВД), катетерного лечения нарушений ритма сердца не выявлено. На фоне регулярного приема медикаментозной терапии отмечалась положительная динамика в виде уменьшения перебоев в работе сердца и одышки при нагрузке. Весной 2021г после перенесенной новой коронавирусной инфекции пациентка вновь отметила нарастание одышки и возобновление аритмичного сердцебиения. По данным контрольного ХМ-ЭКГ (на фоне приема пропафенона 150 мг/сут.) регистрировалась частая ЖЭС с неустойчивыми пробежками желудочковой тахикардии (ЖТ), атриовентрикулярная блокада I степени, эпизоды удлинения интервала QTc максимально до 520 мс. Амбулаторно была скорректирована терапия: пропафенон отменен, увеличена доза метопролола сукцината и валсартана/сакубитрила. В дальнейшем у пациентки сохранялись прежние жалобы, в связи с чем она была направлена в ФГБУ "НМИЦ ТПМ" Минздрава России (г. Москва) для уточнения диагноза.
При лабораторном обследовании основные показатели были в пределах референсных значений, уровень N-концевого промозгового натрийуретического пептида составил 73 пг/мл. По ЭКГ — синусовый ритм с ЧСС 70 уд./мин, электрическая ось сердца расположена горизонтально, одиночная полиморфная ЖЭС, слабо отрицательные зубцы Т в отведениях V4-6, I, aVL, снижение вольтажа в стандартных отведениях. По данным ХМ-ЭКГ (на фоне приема метопролола сукцината 50 мг/сут.) регистрировался синусовый ритм с ЧСС 54-70-142 уд./мин, преходящая атриовентрикулярная блокада I степени, 1423 ЖЭС, 1 неустойчивая пробежка ЖТ. По ЭхоКГ сохранялись незначительная дилатация ЛЖ (конечный диастолический размер 5,3 см), повышенная трабекулярность ЛЖ и правого желудочка (ПЖ) (критерий Jenni), очаги повышенной эхогенности в межжелудочковой перегородке (МЖП), также было описано локальное аневризматическое выпячивание в области верхушки ПЖ. При повторной МРТ сердца с отсроченным внутривенным контрастированием — картина бивентрикулярной некомпактной КМП (критерии Peterson, Jacquier [20]), увеличение индексированных КДО и конечно-систолического объема ЛЖ со снижением ФВ ЛЖ до 43%, неровность боковой стенки ПЖ без признаков дискинезии/акинезии и аневризматического выпячивания, умеренное расширение выносящего тракта ПЖ; при отсроченном контрастировании выявлялись обширные зоны субэпикардиального контрастирования миокарда МЖП, всех сегментов боковой и нижней стенок (рис. 1). По сравнению с данными МРТ от 2020г протяженность зон контрастирования увеличилась. При проведении Т2-картирования признаков отека миокарда отмечено не было, значение параметра ECV (объем внеклеточного матрикса) было повышено до 38% (соответствует наличию фиброза миокарда).
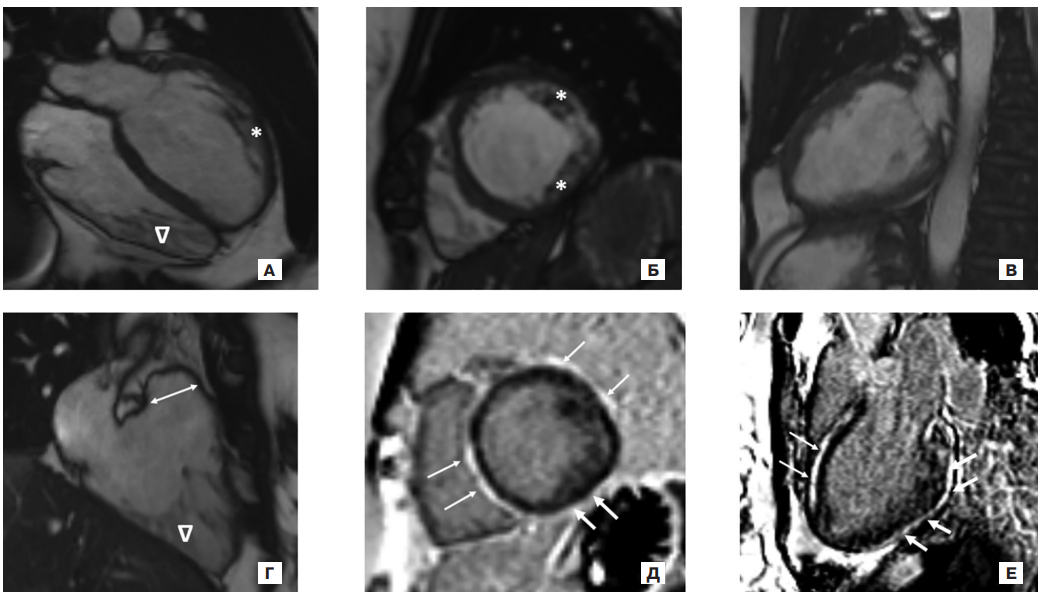
Рис. 1. МРТ сердца пробанда. (а-г) — кино-режим, SSFP-последовательность: а — длинная ось ЛЖ 4-камерная проекция, б — короткая ось на уровне средних сегментов ЛЖ, в — длинная ось ЛЖ 2-камерная проекция, г — длинная ось ПЖ 3-камерная проекция, двойной стрелкой указан выносящий тракт ПЖ (29 мм, несколько расширен); (д, е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда.
Примечание: ЛЖ умеренно расширен (индексированный КДО ЛЖ — 104 мл/м2 при норме 50-96 мл/м2), сократимость его снижена за счет диффузного гипокинеза, ФВ ЛЖ 43%. ПЖ не расширен (индексированный КДО ПЖ — 78 мл/м2 при норме 48-87 мл/м2), сократимость его не снижена. Гипертрофии миокарда нет (индексированная масса миокарда ЛЖ — 57 г/м2 при норме 32-66 г/м2). Трабекулярность миокарда ЛЖ и ПЖ повышена. * — повышенная трабекулярность по боковой стенке ЛЖ за счет "рассыпного" типа строения папиллярных мышц, ∆ — повышенная трабекулярность в верхушечной области ПЖ. Стрелками указаны протяженные зоны субэпикардиального контрастирования по боковой и нижней стенкам ЛЖ и в МЖП со стороны полости ПЖ.
Сокращения: КДО — конечно-диастолический объем, ЛЖ — левый желудочек, МЖП — межжелудочковая перегородка, ПЖ — правый желудочек, ФВ — фракция выброса.
Пробанду было проведено молекулярно-генетическое исследование. По результатам секвенирования следующего поколения был выявлен гетерозиготный патогенный вариант в гене DSP (hg19::chr6:7577240del, ENST00000379802.3:c.2842del, ENSP00000369129.3:p. Gln948LysfsTer29) [17], который был подтвержден секвенированием по Сенгеру.
Учитывая выявленные желудочковые нарушения ритма сердца, отягощенный семейный анамнез (описан далее), распространенный субэпикардиальный фиброз, не соответствующий выраженности дилатации и дисфункции ЛЖ, патогенный вариант в АКМП-ассоциированном гене (DSP), на основании обновленных Падуанских критериев АКМП от 2020 [21][22] пациентке был выставлен диагноз: "Генетически детерминированная КМП (патогенный вариант в гене DSP): аритмогенная левожелудочковая КМП, некомпактный миокард, бивентрикулярная форма".
В стационаре оптимизирована антиаритмическая терапия (увеличена доза метопролола сукцината до 75 мг/сут.) и терапия хронической СН (инициирован прием дапаглифлозина, продолжен прием сакубитрила/валсартана, эплеренона).
С учетом высокого риска развития жизнеугрожающих желудочковых аритмий, ассоциированных с патогенным вариантом гена DSP и выраженным фиброзом, с целью первичной профилактики ВСС было принято решение об имплантации КВД в плановом порядке. Также пациентке были даны рекомендации по ограничению физической нагрузки.
Фенотипический каскадный скрининг
В рамках семейного скрининга были обследованы мать и дети пробанда.
Мать пробанда, 63 лет, в возрасте 56 лет (2015г) перенесла эпизод интенсивной загрудинной боли и слабости, однако за медицинской помощью не обратилась. В 2019г в возрасте 60 лет впервые на ЭхоКГ был выявлен диффузный гипокинез миокарда ЛЖ, акинез передней стенки ЛЖ со снижением ФВ ЛЖ до 34%. Данные изменения были расценены как постинфарктный кардиофиброз, инициирована терапия бета-адреноблокаторами и спиронолактоном. Пациентка чувствовала себя удовлетворительно вплоть до весны 2022г, когда, в связи с нарастанием жалоб на одышку при физической нагрузке, отеки нижних конечностей и увеличение живота в объеме, была госпитализирована в стационар по месту жительства. По ЭхоКГ при поступлении описана дилатация всех камер сердца, диффузный гипокинез миокарда ЛЖ со снижением ФВ ЛЖ до 20%, легочная гипертензия, митральная регургитация 2 степени, трикуспидальная регургитация 3 степени, также впервые выявлены признаки некомпактности миокарда. Во время госпитализации у пациентки развился устойчивый пароксизм ЖТ, сопровождавшийся гемодинамической нестабильностью и потерей сознания, в связи с чем экстренно проведена электроимпульсная терапия с восстановлением синусового ритма. По данным ХМ-ЭКГ после кардиоверсии регистрировался синусовый ритм с ЧСС 71-80-101 уд./мин, 2000 ЖЭС, ЖТ не было. Проведена коронарография, по данным которой коронарные артерии без гемодинамически значимых стенозов. Инициирована комплексная терапия СН, с антиаритмической целью назначен амиодарон в комбинации с бисопрололом. В дальнейшем с целью вторичной профилактики ВСС была выполнена имплантация КВД. На фоне проводимой терапии отмечалась положительная динамика в виде компенсации явлений СН, срабатываний КВД зафиксировано не было.
Детям пробанда — старшая дочь 19 лет (профессионально занимается волейболом), младшая дочь 14 лет, сын 11 лет — проведено комплексное кардиологическое обследование (ЭКГ, ЭхоКГ, ХМ-ЭКГ), по результатам которого патологии не выявлено.
Бабушка пробанда по материнской линии скончалась в возрасте 54 лет от острого нарушения мозгового кровообращения.
Дедушка пробанда по материнской линии внезапно скончался в возрасте 52 лет.
Данные молекулярно-генетической диагностики
Родственникам первой степени родства было проведено секвенирование по Сенгеру для определения наличия варианта в гене DSP, выявленного у пробанда. Вариант DSP: p.Gln948LysfsTer29 в гетерозиготном состоянии был выявлен у матери и старшей дочери пробанда.
Десмоплакиновая КМП — новый термин, предложенный для описания особой формы КМП, отличающейся от общепринятых классических фенотипов [8][9]. Фенотипически DSP-ассоциированная КМП является неоднородным заболеванием и может быть отнесена как к АКМП (в доминирующем числе случаев — левожелудочковой форме, реже — правожелудочковой форме), так и к ДКМП, и некомпактной КМП [12-14]. В то же время, несмотря на морфологическую гетерогенность, десмоплакиновая КМП характеризуется рядом специфических черт, которые определяют ее плохой прогноз и, следовательно, клиническую значимость как самостоятельной нозологии.
Основным клиническим проявлением десмоплакиновой КМП являются желудочковые нарушения ритма, такие как частая ЖЭС (>500/сут.), неустойчивые и устойчивые ЖТ (обычно с паттерном блокады правой ножки пучка Гиса), а также фибрилляция желудочков. Предрасположенность к тяжелым аритмиям ранее была описана Smith ED, et al. (2020) у 28% пациентов с вариантами в гене DSP, причем особенно высокая частота встречаемости наблюдалась у пробандов (43%) [8]. Аналогично в исследовании Wang W, et al. (2022) отмечена высокая частота злокачественных желудочковых аритмий (27%), из которых значительная часть была представлена фибрилляцией желудочков/прерванной ВСС (17%) [9].
Другой отличительной чертой десмоплакиновой КМП является распространенный фиброз миокарда ЛЖ, который лежит в основе патологического процесса и предшествует развитию дилатации и дисфункции ЛЖ. Учитывая, что фиброзные изменения обычно ограничиваются внешними слоями сердечной стенки, систолическая функция ЛЖ длительное время значительно не страдает. Этот факт объясняет ограниченность применения ЭхоКГ для выявления субэпикардиального поражения ЛЖ, поскольку нарушения локальной и глобальной сократимости, а также дилатация ЛЖ могут отсутствовать вплоть до поздних стадий заболевания за счет сохранного субэндокардиального и среднего слоев миокарда [20]. Основным методом визуализации при диагностике десмоплакиновой КМП является МРТ с контрастным усилением гадолинием, для которого в отсроченную фазу характерно субэпикардиальное накопление контрастного препарата с формированием линейных и кольцевидных сливных зон, распространяющихся более чем на два сегмента ЛЖ [8][9].
Ещё одной специфической клинической особенностью DSP-ассоциированной КМП является возникновение рецидивирующих эпизодов миокардиального повреждения, характеризующихся болью в груди в сочетании с документально подтвержденным поражением миокарда (повышение уровня маркеров повреждения миокарда при неизмененных коронарных артериях) [15, 16]. По литературным данным у пациентов с десмоплакиновой КМП частота встречаемости таких миокардит-подобных эпизодов варьирует от 14 до 22% [8][9]. В недавнем многоцентровом исследовании проводилось сравнение результатов лечения пациентов с острым миокардитом в зависимости от наличия у них вариантов в десмосомальных генах. У пациентов с верифицированными патогенными вариантами (89% с вариантами в гене DSP) наблюдалась значительно более высокая частота достижения основной конечной точки (смерти, желудочковых аритмий, рецидивирующих эпизодов миокардита и СН), чем у лиц без патогенных вариантов (62,3% против 17,5% в течение 5 лет, р<0,0001) [23][24]. Учитывая эти данные, в настоящее время предполагается, что эпизоды острого воспалительного повреждения миокарда могут играть роль в патогенезе развития фиброза и дисфункции ЛЖ у пациентов с вариантами в гене DSP [9].
Представленное в нашей работе клиническое наблюдение наглядно демонстрирует гетерогенность проявлений десмоплакиновой КМП на разных этапах её развития. Так, у пробанда отмечается смешанный фенотип, характеризующийся высокой аритмогенностью, умеренной систолической дисфункцией и незначительной дилатацией ЛЖ в сочетании с признаками бивентрикулярной некомпактности, что, согласно современной классификации, соответствует левожелудочковой АКМП и некомпактной КМП. В свою очередь, у матери пробанда в настоящий момент наблюдается выраженная дилатация камер сердца со значимым снижением глобальной сократимости ЛЖ и также имеются критерии некомпактного миокарда, что позволяет отнести данный фенотип к категориям ДКМП и некомпактной КМП. Однако как у пробанда, так и у матери пробанда присутствует отличительный признак десмоплакиновой КМП — распространенный субэпикардиальный фиброз миокарда ЛЖ, который лежит в основе развития КМП и выступает в качестве первичного субстрата для желудочковых аритмий. В совокупности это позволяет сделать вывод о том, что описанные фенотипы, наиболее вероятно, являются стадиями одного патологического процесса, а не отдельными формами КМП. Так, на примере описанной семьи можно проследить этапы естественного течения десмоплакиновой КМП: 1) доклиническая стадия — предположительно, стадия старшей дочери пробанда, учитывая наличие подтвержденного патогенного варианта гена DSP и отсутствие структурной и функциональной патологии сердца по данным проведенного обследования; 2) пресимптоматическая/малосимптомная стадия, когда появляются электрические (изменения ЭКГ, ЖЭС, неустойчивые ЖТ), а затем структурные нарушения (дилатация, глобальная и/или локальная дисфункция желудочков) без выраженных специфических проявлений — стадия пробанда; 3) клиническая стадия, которая характеризуется приступами ЖТ, синкопе и ВСС — стадия матери пробанда [25]. Именно наличие бессимптомной стадии диктует необходимость проведения обязательного генетического каскадного скрининга семьи после идентификации заболевания у пробанда, что важно для ранней диагностики и своевременного принятия мер с целью профилактики ВСС.
Учитывая относительно недавнее выделение десмоплакиновой КМП в отдельную нозологическую единицу [8][9], сведения о возможностях таргетной терапии в настоящее время отсутствуют, и лечение ограничивается коррекцией СН, нарушений ритма сердца и профилактикой ВСС. Вопрос о стратификации риска ВСС также остается неоднозначным. Попытка адаптировать уже существующие критерии оценки риска, разработанные для классической правожелудочковой АКМП и ДКМП, показала их недостаточную прогностическую точность в случае десмоплакиновой КМП. Так, например, в исследовании Smith ED, et al. (2020) стандартное пороговое значение ФВ ЛЖ <35% при ДКМП не было чувствительным маркером аритмических событий в когорте пациентов с вариантами в гене DSP, не позволив выявить 52% пациентов с жизнеугрожающими нарушениями ритма [8]. Учитывая выраженность фиброза, лежащего в основе желудочковых аритмий, но затрагивающего преимущественно субэпикардиальные слои миокарда без развития значимой систолической дисфункции, по данным многих авторов следует использовать в качестве дополнительного критерия стратификации риска более высокий уровень ФВ ЛЖ <55% [8][23]. Аналогичным образом, классические факторы риска, учитываемые при правожелудочковой АКМП, такие как снижение сократительной способности ПЖ, инверсия зубца Т на ЭКГ и мужской пол, не были прогностически значимыми в оценке аритмического риска при десмоплакиновой КМП [8][9]. Исходя из предполагаемых патогенетических механизмов, вероятно, наиболее чувствительными маркерами риска могут являться преобладающее поражение ЛЖ, рецидивирующие эпизоды острого повреждения миокарда, обширный фиброз по данным МРТ сердца с контрастированием и наличие частой ЖЭС и неустойчивых ЖТ по данным суточного мониторирования ЭКГ, однако для уточнения факторов риска ВСС и разработки четких критериев имплантации КВД требуются дальнейшие исследования на более крупных когортах пациентов с вариантами в гене DSP. В настоящее время однозначным остается лишь необходимость генотип-специфического подхода, нашедшая отражение и в недавно опубликованных клинических рекомендациях Европейского общества кардиологов по лечению КМП, в которых варианты гена DSP выделены в категорию высокого аритмического риска с такими дополнительными предикторами, как позднее накопление гадолиния при МРТ сердца и ФВ ЛЖ <45% [7]. Как было отмечено ранее, МРТ с мультипараметрическим картированием в настоящее время является "золотым стандартом" неинвазивной характеристики состояния миокарда ЛЖ. Выявление признаков фиброзных изменений миокарда по данным МРТ является неблагоприятным прогностическим фактором развития таких осложнений, как СН и жизнеугрожающие нарушения ритма сердца. Кроме того, учитывая имеющиеся на сегодняшний день данные о роли воспалительного поражения миокарда у пациентов с десмоплакиновой КМП, на наш взгляд целесообразно в ходе рутинной МРТ сердца проводить нативное Т2-картирование для выявления отека и количественное определение объема внеклеточного матрикса (ECV) для оценки прогноза. В нашем клиническом наблюдении, согласно современной концепции, пробанду была рекомендована имплантация КВД, несмотря на умеренную систолическую дисфункцию ЛЖ.
Также неопределенной при десмоплакиновой КМП остается проблема допустимой физической активности. Ранее в ряде исследований было показано, что интенсивность физических упражнений была напрямую связана с прогрессированием СН и повышением риска возникновения жизнеугрожающих аритмий при "классических" правожелудочковых формах АКМП [26], однако в изучаемых когортах были незначительно представлены пациенты с преобладающим поражением ЛЖ, что не позволяет транслировать полученные результаты на пациентов с вариантами в гене DSP. Согласно данным Smith ED, et al. (2020), напротив, различий в степени систолической дисфункции и аритмической нагрузки между группами с разным уровнем активности получено не было [8], но этих результатов недостаточно для создания рекомендаций относительно объема физических упражнений, и необходимы дальнейшие исследования в этой сфере. В представленном клиническом случае, в связи с выявленным патогенным вариантом в гене DSP, старшей дочери пробанда в настоящее время было рекомендовано проведение МРТ сердца с целью исключения начальных фиброзных изменений миокарда ЛЖ для решения вопроса о необходимости ограничения интенсивных тренировок, учитывая занятия профессиональным спортом.
Десмоплакиновая КМП — уникальная форма КМП, характеризующаяся распространенным фиброзом ЛЖ, жизнеугрожающими желудочковыми аритмиями и эпизодами воспалительного повреждения миокарда. Выделение десмоплакиновой КМП подтверждает, что в перспективе применение генотип-специфического подхода к диагностике и классификации КМП будет способствовать совершенствованию методов стратификации риска и выбора тактики ведения пациентов в эпоху развивающейся персонализированной медицины.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-6. doi:10.1093/eurheartj/ehm342.
2. Paldino A, Dal Ferro M, Stolfo D, et al. Prognostic Prediction of Genotype vs Phenotype in Genetic Cardiomyopathies. J Am Coll Cardiol. 2022;80(21):1981-94. doi:10.1016/j.jacc.2022.08.804.
3. Van Tintelen J, Entius M, Bhuiyan Z, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113(13):1650-8. doi:10.1161/CIRCULATIONAHA.105.609719.
4. Gigli M, Stolfo D, Graw S, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation. 2021;144(20):1600-11. doi:10.1161/CIRCULATIONAHA.121.053521.
5. Hasselberg N, Haland T, Saberniak J, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. 2018;39(10):853- 60. doi:10.1093/eurheartj/ehx596.
6. Akhtar M, Lorenzini M, Cicerchia M, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN Gene. Circ Heart Fail. 2020;13(10):e006832. doi:10.1161/CIRCHEARTFAILURE.119.006832.
7. Arbelo E, Protonotarios A, Gimeno J, et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur Heart J. 2023;44:3503-626. doi:10.1093/eurheartj/ehad194.
8. Smith E, Lakdawala N, Papoutsidakis N, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1872-84. doi:10.1161/CIRCULATIONAHA.119.044934.
9. Wang W, Murray B, Tichnell C, et al. Clinical Characteristics and Risk Stratification of Desmoplakin Cardiomyopathy. EP Eur. 2022;24:268-77. doi:10.1093/europace/euab183.
10. Yuan Z, Cheng L, Wang Z, et al. Desmoplakin and Clinical Manifestations of Desmoplakin Cardiomyopathy. Chin. Med. J. 2021;134:1771-9. doi:10.1097/CM9.000000000000158.
11. Norgett E, Hatsell S, Carvajal-Huerta L, et al. Recessive Mutation in Desmoplakin Disrupts Desmoplakin-Intermediate Filament Interactions and Causes Dilated Cardiomyopathy, Woolly Hair and Keratoderma. Hum. Mol. Genet. 2000;9:2761-6. doi:10.1093/hmg/9.18.2761.
12. Corrado D, Basso C. Arrhythmogenic left ventricular cardiomyopathy. Heart. 2022;108:733-43. doi:10.1136/heartjnl-2020-316944.
13. López-Ayala J, Gómez-Milanés I, Sánchez Muñoz J, et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. Europace. 2014;16(12):1838-46. doi:10.1093/europace/euu128.
14. Augusto J, Eiros R, Nakou E, et al. Dilated Cardiomyopathy and Arrhythmogenic Left Ventricular Cardiomyopathy: A Comprehensive Genotype-Imaging Phenotype Study. Eur Heart J.Cardiovasc. Imaging. 2020;21:326-36. doi:10.1093/ehjci/jez188.
15. Ghawanmeh M, Simon Frances B, Kerai A, et al. Management of Recurrent Myocarditis Due to Desmoplakin Cardiomyopathy: Diagnostic and Therapeutic Challenges. JACC Case Rep. 2022;4:59-62. doi:10.1016/j.jaccas.2021.10.005.
16. Rezaei Bookani K, Minga I, Wodskow J, et al. A case series of desmoplakin cardiomyopathy: A mimic of viral myocarditis. Eur Heart J.Case Rep. 2022;6:ytac341. doi:10.1093/ehjcr/ytac341.
17. Meshkov AN, Myasnikov RP, Kiseleva AV, et al. Genetic landscape in Russian patients with familial left ventricular noncompaction. Frontiers in Cardiovascular Medicine. 2023;10:1205787. doi:10.3389/fcvm.2023.1205787.
18. Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666-71. doi:10.1136/heart.86.6.666.
19. Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46(1):101-5. doi:10.1016/j.jacc.2005.03.045.
20. Jacquier A, Thuny F, Jop B, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J. 2010;31(9):1098-104. doi:10.1093/eurheartj/ehp595.
21. Corrado D, Perazzolo Marra M, Zorzi A, et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J.Cardiol. 2020;319:106-14. doi:10.1016/j.ijcard.2020.06.005.
22. Вайханская Т. Г., Сивицкая Л. Н., Курушко Т. В. и др. Смена концепции аритмогенной кардиомиопатии: расширение клиникогенетического спектра, новые критерии диагностики левожелудочковых фенотипов. Российский кардиологический журнал. 2020;25(10):3863. doi:10.15829/1560-4071-2020-3863.
23. Cipriani A, Bauce B, De Lazzari M, et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis with Dilated Cardiomyopathy. J.Am. Heart Assoc. 2020;9:e014628. doi:10.1161/JAHA.119.014628.
24. Ammirati E, Raimondi F, Piriou N, et al. Acute Myocarditis Associated with Desmosomal Gene Variants. JACC Heart Fail. 2022;10:714-27. doi:10.1016/j.jchf.2022.06.013.
25. Mattesi G, Zorzi A, Corrado D, et al. Natural History of Arrhythmogenic Cardiomyopathy. J Clin Med. 2020;9(3):878-92. doi:10.3390/jcm9030878.
26. Bosman L, Wang W, Lie Ø, et al. Integrating Exercise Into Personalized Ventricular Arrhythmia Risk Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Arrhythm Electrophysiol. 2022;15(2):e010221. doi:10.1161/CIRCEP.121.010221.
Кандидат медицинских наук, руководитель лаборатории персонализированной диагностики, профилактики и терапии некоронарогенных заболеваний сердца Института персонализированной терапии и профилактики, ведущий научный сотрудник отдела клинической кардиологии
Москва
Лаборант-исследователь отдела клинической кардиологии
Москва
Лаборант-исследователь отдела клинической кардиологии
Москва
Кандидат биологических наук, руководитель лаборатории молекулярной генетики Института персонализированной терапии и профилактики
Москва
Кандидат медицинских наук, старший научный сотрудник лаборатории персонализированной диагностики, профилактики и терапии некоронарогенных заболеваний сердца Института персонализированной терапии и профилактики, отдела клинической кардиологии
Москва
Доктор медицинских наук, руководитель Института персонализированной терапии и профилактики, руководитель отдела персонализированной диагностики, профилактики и терапии атеросклеротических сердечно-сосудистых заболеваний
Москва
Младший научный сотрудник лаборатории персонализированной диагностики, профилактики и терапии некоронарогенных заболеваний сердца Института персонализированной терапии и профилактики, отдела клинической кардиологии
Москва
Кандидат медицинских наук, доцент кафедры лучевой диагностики и лучевой терапии, зав. отделением рентгенодиагностики с кабинетами магнитно-резонансной и компьютерной томографии
Москва
Кандидат биологических наук, старший научный сотрудник лаборатории молекулярной генетики Института персонализированной терапии и профилактики ФГБУ Национальный медицинский исследовательский центр профилактической медицины Минздрава России; доцент кафедры генетики, биотехнологии ФГБНУ Всероссийский научно-исследовательский институт сельскохозяйственной биотехнологии
Москва
Младший научный сотрудник лаборатории персонализированной диагностики, профилактики и терапии некоронарогенных заболеваний сердца Института персонализированной терапии и профилактики, отдела клинической кардиологии ФГБУ Национальный медицинский исследовательский центр терапии и профилактической медицины Минздрава России, ассистент кафедры лучевой диагностики факультета фундаментальной медицины ФГБОУ ВО МГУ им. М.В. Ломоносова
Москва
Кандидат медицинских наук, ведущий научный сотрудник отдела нарушений сердечного ритма и проводимости сердца
Москва
Академик РАН, доктор медицинских наук, профессор, директор
Москва
Мясников Р.П., Кузина Н.Н., Нефедова Д.А., Киселева А.В., Куликова О.В., Мешков А.Н., Кудрявцева М.М., Мершина Е.А., Дивашук М.Г., Рыжкова Е.В., Харлап М.С., Драпкина О.М. Десмоплакин и особенности течения десмоплакиновой кардиомиопатии. Российский кардиологический журнал. 2023;28(11):5648. https://doi.org/10.15829/1560-4071-2023-5648. EDN: JSXHYA
Myasnikov R.P., Kuzina N.N., Nefedova D.A., Kiseleva A.V., Kulikova O.V., Meshkov A.N., Kudryavtseva M.M., Mershina E.A., Divashuk M.g., Ryzhkova E.V., Kharlap M.S., Drapkina O.M. Desmoplakin and features of desmoplakin cardiomyopathy. Russian Journal of Cardiology. 2023;28(11):5648. (In Russ.) https://doi.org/10.15829/1560-4071-2023-5648. EDN: JSXHYA













































