

Содержание
Перейти к:
 Д. С. Бутенко,
Р. И. Алекберов,
В. В. Спасенников,
Т. М. Клещевникова,
А. Ю. Талыбова,
Р. С. Талыбов
Д. С. Бутенко,
Р. И. Алекберов,
В. В. Спасенников,
Т. М. Клещевникова,
А. Ю. Талыбова,
Р. С. Талыбов https://doi.org/10.15829/1560-4071-2025-6425
EDN: OUXIDK
Перейти к:
Введение. Синдром Шерешевского-Тернера (сШ-Т) представляет собой генетическую патологию, обусловленную полной или частичной моносомией по X-хромосоме. Для данного заболевания характерен полиморфизм врожденных пороков развития, с преимущественным вовлечением эндокринной и сердечно-сосудистой систем. Пациенты с сШ-Т демонстрируют более высокие показатели заболеваемости и смертности в сравнении с общей популяцией, что диктует необходимость комплексного междисциплинарного подхода к их ведению.
Краткое описание. В статье представлен клинический случай пациентки с генетически подтвержденным сШ-Т (кариотип 45,X), у которой прижизненно диагностировано осложнение — расслоение аорты (тип III по Дебейки), что стало возможным благодаря комплексному обследованию. В работе детально описаны характерные фенотипические проявления, особенности клинической картины и течения заболевания, анализ факторов риска, примененные методы лабораторной и инструментальной диагностики и подходы к лечебной тактике.
Дискуссия. Ключевой задачей ведения пациентов с врожденными генетическими заболеваниями соединительной ткани является поддержание высокого уровня клинической настороженности среди врачей всех специальностей. Критически важным представляется не только осознание специфических рисков, ассоциированных с генетическим синдромом, но и тщательная оценка потенциальных триггерных факторов, способных спровоцировать острое сосудистое событие. Оперативное предположение и верификация жизнеугрожающих состояний, к числу которых относится расслоение аорты, определяют выбор неотложной лечебной тактики и могут предотвратить летальный исход.
Бутенко Д.С., Алекберов Р.И., Спасенников В.В., Клещевникова Т.М., Талыбова А.Ю., Талыбов Р.С. Расслоение аорты у пациента с синдромом Шерешевского-Тернера. Клинический случай. . 2025;30(10S):6425. https://doi.org/10.15829/1560-4071-2025-6425. EDN: OUXIDK
Butenko D.S., Alekberov R.I., Spasennikov V.V., Kleshchevnikova T.M., Talybova A.Yu., Talybov R.S. Aortic dissection in a patient with Turner syndrome: a case report. . 2025;30(10S):6425. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6425. EDN: OUXIDK
Синдром Шерешевского-Тернера (сШ-Т) — распространенное хромосомное заболевание, обусловленное полной или частичной моносомией Х-хромосомы [1]. Классический фенотип (кариотип 45,X) характеризуется низким ростом, дисфункцией гонад и врожденными сердечно-сосудистыми пороками, включая коарктацию аорты (КоА) и двустворчатый аортальный клапан (ДАК) [2]. Одним из важнейших аспектов является значительное сокращение ожидаемой продолжительности жизни у пациенток (в среднем на 12,5 лет), обусловленное преимущественно сердечно-сосудистыми осложнениями [3]. Данные международных регистров свидетельствуют о 3-кратном повышении общей смертности при сШ-Т, причем сердечно-сосудистые заболевания являются ведущей причиной смерти (до 50% случаев) [4].
Расслоение аорты (РА) представляет собой критическое, жизнеугрожающее состояние с высокой ранней летальностью (до 21% в первые 24 ч) и сложностями диагностики (85% первоначальных ошибок из-за неспецифичности болевого синдрома) [4]. сШ-Т является наиболее частой установленной причиной РА у молодых женщин. Риск РА при сШ-Т значительно повышен во всех возрастных группах, достигая максимума в молодом возрасте и во время беременности. В отличие от общей популяции, где РА чаще встречается у мужчин старше 65 лет, у пациенток с сШ-Т это осложнение развивается необычно рано (часто до 40 лет). Помимо специфического поражения аорты (удлинение дуги, аневризмы, коарктация, расслоение, разрыв, клапанные аномалии), при сШ-Т наблюдается системная васкулопатия с возможным вовлечением магистральных артерий головы и периферических сосудов [5].
Основными факторами риска РА при сШ-Т являются КоА, ДАК, артериальная гипертензия (АГ) и расширение восходящей аорты. Для стратификации риска используется индекс размера аорты (aortic size index, ASI), рассчитываемый с поправкой на площадь поверхности тела [6]. Международные рекомендации регламентируют рассматривать профилактическую хирургическую коррекцию при ASI ≥2,5 см/м2 восходящего отдела аорты у пациенток старше 15 лет [6]. Однако данный порог имеет существенные ограничения: он основан на слабой доказательной базе (отсутствуют проспективные исследования, специфичные для сШ-Т), зафиксированы случаи РА у пациенток с ASI ниже этого значения, а сама профилактическая операция сопряжена с риском летальности до 5% [7][8]. Это создает серьезную клиническую дилемму при выборе тактики ведения: необходимость баланса между риском фатального РА при выжидательной тактике и риском неоправданного хирургического вмешательства. Таким образом, сШ-Т ассоциирован с уникально высоким риском раннего и жизнеугрожающего РА, являющегося ведущей причиной сокращения продолжительности жизни пациенток [9].
Моногенные заболевания соединительной ткани, включая сШ-Т, представляют собой противопоказание к транскатетерной изоляции РА ввиду высокого риска расширения ложного просвета и несостоятельности фиксации стент-графта. Тем не менее в критических неотложных ситуациях данная методика может рассматриваться в качестве компромиссного решения. Согласно консенсусным рекомендациям Общества сосудистой хирургии (Society for Vascular Surgery) и Общества торакальных хирургов (Society of Thoracic Surgeons), Национальным клиническим рекомендациям и данным современных обзоров литературы, эндоваскулярное вмешательство допустимо лишь при абсолютной невозможности выполнения открытой операции, наличии выраженной ишемии висцеральных органов или нестабильной гемодинамики при условии отсутствия альтернативных терапевтических решений [10][11]. Представлены единичные случаи успешного лечения РА методом транскатетерной изоляции области расслоения стент-графтом, однако количество опубликованных клинических наблюдений остается ограниченным на текущий момент и требует дальнейшего изучения [12][13].
Отсутствие подтвержденных и надежных критериев для принятия решения о профилактической операции на аорте (в частности, неоднозначность порога ASI ≥2,5 см/м2) подчеркивает актуальность дальнейших исследований для оптимизации стратегий наблюдения, стратификации риска и профилактического лечения этой уязвимой группы пациентов. Нами представлен клинический случай РА тип 3 по классификации DeBakey у молодой женщины 32 лет с сШ-Т, демонстрирующий практическую значимость данной проблемы.
Пациентка М., 32 лет, поступила в приемное отделение ГБУЗ ТО "ОКБ № 2" с жалобами на болевой синдром в грудной клетке, преимущественно в межлопаточной области. Данные центральной электронной медицинской карты свидетельствуют о неоднократных обращениях пациентки в поликлинику по месту жительства на протяжении предшествующего месяца по поводу болей в пояснице, спине и грудной клетке. Учитывая выраженный психоэмоциональный фон пациентки и волнообразное течение симптоматики (самопроизвольное возникновение и исчезновение болевого синдрома), на амбулаторном этапе состояние было интерпретировано как проявление вегетососудистой дистонии. Пациентке было рекомендовано амбулаторное лечение. В связи с персистенцией симптомов она самостоятельно обратилась в приемное отделение стационара экстренного профиля.
Ввиду локализации болевого синдрома пациент проконсультирован врачом-кардиологом. При осмотре обращала на себя внимание нетипичная морфология пациентки: низкий рост (145 см), масса тела 70 кг (индекс массы тела 33 кг/м2), широкая грудная клетка и крыловидная шея. Физикальное обследование значимых отклонений не выявило. Артериальное давление (АД) на момент осмотра составляло 160/80 мм рт.ст. Межлопаточная боль не усиливалась при пальпации паравертебральных мышц и остистых отростков позвоночника, что снижало вероятность ее связи с дегенеративными изменениями позвоночного столба.
Пациентке выполнена электрокардиография и забор крови для лабораторных исследований, включавших: общий анализ крови, биохимический анализ крови (С-реактивный белок, креатинфосфокиназа, глюкоза, мочевина, альбумин, хлор, натрий, калий, креатинин, билирубин прямой, билирубин общий, аспартатаминотрансфераза, аланинаминотрансфераза), тропонин I, коагулограмму (активированное частичное тромбопластиновое время, международное нормализованное отношение, протромбиновое время, фибриноген). В период ожидания результатов отмечалось усиление болевого синдрома. Выраженность болей в сочетании с нехарактерной внешностью вызвали подозрение на сШ-Т и, с учетом клинической картины, на возможный факт РА.
При повторном сборе анамнеза пациентка подтвердила наличие у себя данного генетического заболевания. Повышение АД отмечалось в течение последнего месяца, что пациентка связывала с диагностированным ранее манифестным гипотиреозом (уровень тиреотропного гормона достигал 10,7 мМЕ/л) и приемом L-тироксина в дозе 125 мкг/сут. Было принято решение о дополнительном назначении определения концентрации D-димера. По результатам обследований выявлено повышение фибриногена (4,44 г/л) и D-димера (4,001 мг/л). Остальные лабораторные показатели и тропонин I находились в пределах референсных значений.
На электрокардиографии: синусовый ритм с частотой сердечных сокращений 73 уд./мин, неполная блокада правой ножки пучка Гиса. Пациентка была направлена на компьютерную томографию (КТ) органов грудной полости с контрастированием. КТ-ангиография визуализировала дефект контрастирования аорты от уровня дуги до уровня Th12 позвонка. Истинный просвет аорты составлял 1,0 см с сужением в дистальных отделах. На основании рентгенологического исследования сформулировано предположение о РА типа III по классификации DeBakey.
По решению врачебного консилиума для верификации диагноза выполнены КТ-панаортография и эхокардиография. Данные КТ-панаортографии подтвердили наличие расслоения, протянувшегося от уровня, расположенного дистальнее отхождения левой подключичной артерии, и до инфраренального сегмента брюшной аорты. Сопутствующей находкой явилась подковообразная почка с очагом инфаркта в паренхиме правой половины почки (рис. 1, 2).
Результаты эхокардиографии подтвердили расслоение стенки между истинным и ложным просветами нисходящей грудной аорты. Пациентка была осмотрена мультидисциплинарной бригадой (в составе кардиолога, сердечно-сосудистого хирурга, анестезиолога-реаниматолога) в условиях реанимационного зала. На фоне дообследования АД повышалось до 160/90 мм рт.ст., сопровождаясь усилением болевого синдрома. Проведена экстренная коррекция АД: внутривенно введен раствор метопролола (1 мг/мл) в объеме 2 мл, что привело к снижению АД до 110/80 мм рт.ст. в течение 5 минут и купированию боли. После консультации по линии центра медицины катастроф с дежурным сердечно-сосудистым хирургом и компенсации состояния было принято решение о переводе пациентки в профильное кардиохирургическое отделение для решения вопроса о транскатетерной изоляции расслоения стент-графтом. В ходе предоперационной подготовки у пациентки развились критические нарушения гемодинамики. Проведенные реанимационные мероприятия оказались неэффективными, был зарегистрирован летальный исход.
Временная шкала
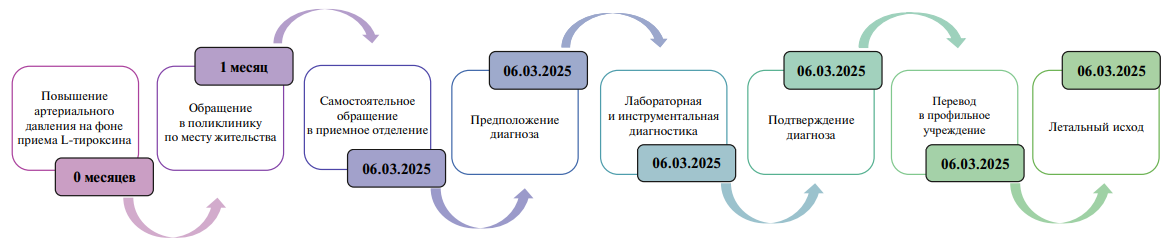
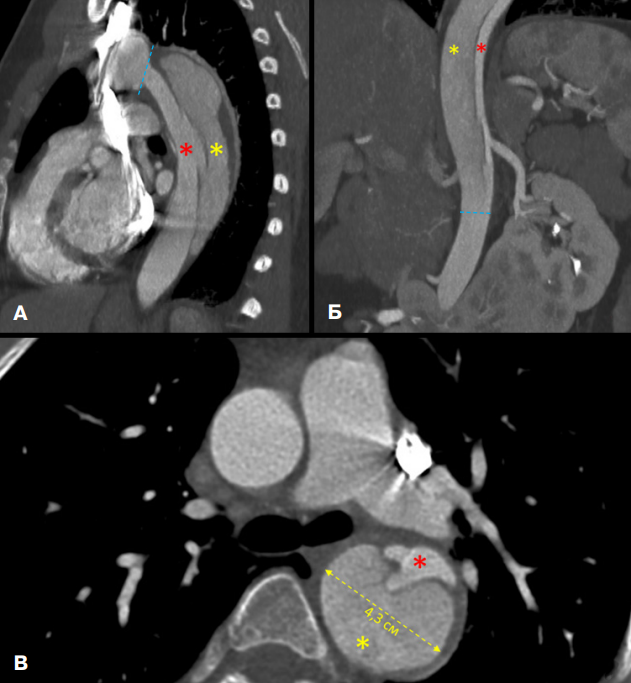
Рис. 1. КТ-панаортография пациентки с РА тип III (De Bakey).
Примечание: А — сагиттальная плоскость, Б — фронтальная плоскость, В — аксиальная плоскость. Проксимальная граница расслоения расположена непосредственно после отхождения левой подключичной артерии от дуги аорты, дистальная граница — на уровне инфраренального отдела брюшного отдела аорты (голубые пунктирные линии). Ложный просвет (желтая звезда) имеет больший размер, чем истинный просвет (красная звезда), наибольший диаметр аорты на уровне нисходящей части — 4,3 см. Четко визуализируется только дистальный разрыв интимы в области дистальной границы. Цветное изображение доступно в электронной версии журнала.
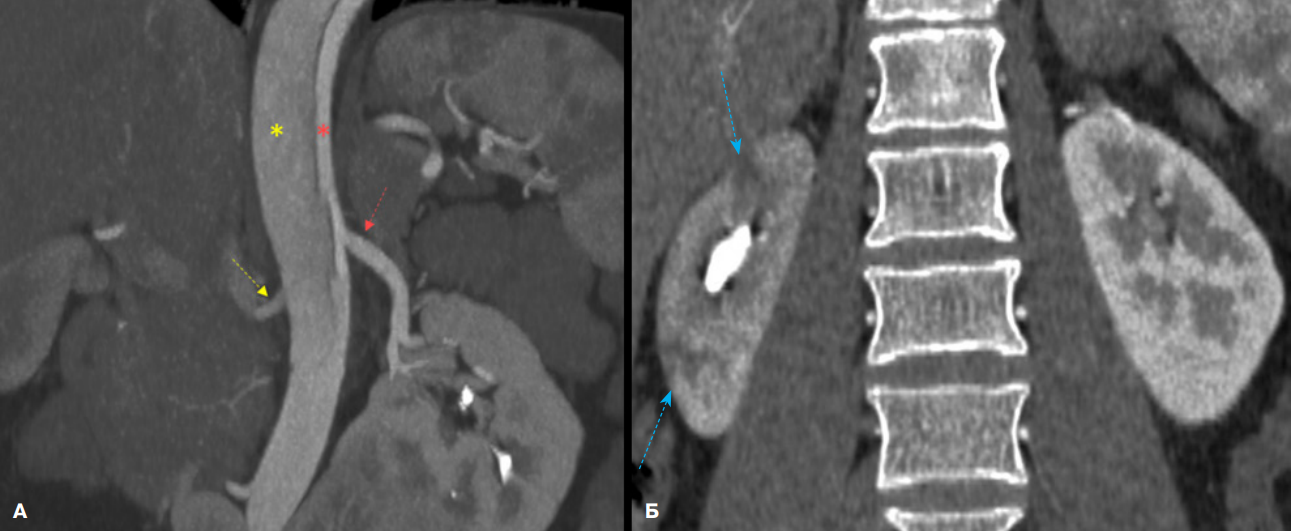
Рис. 2. КТ-панаортография пациента с РА тип III (De Bakey).
Примечание: А — фронтальная плоскость (на уровне отхождения почечных артерий), Б — фронтальная плоскость (на уровне почек). Правая почечная артерия (желтая стрелка) отходит из ложного просвета, в отличии от левой почечной артерии (красная стрелка). Имеются различия общей перфузии паренхимы почек за счёт снижения контрастирования правой почки с формированием инфарктов (синие стрелки), а также слияние нижних полюсов почек (подковообразная почка). Цветное изображение доступно в электронной версии журнала.
Представленное клиническое наблюдение подчеркивает критическую важность комплексного диагностического подхода при оценке острых болевых синдромов у пациенток с сШ-Т. Хотя РА часто ассоциировано с пожилым возрастом, АГ и атеросклерозом, врожденная неполноценность соединительной ткани при сШ-Т существенно снижает возраст манифестации и отягощает течение васкулопатии [14][15]. сШ-Т характеризуется спектром соматических аномалий, включая системную дисплазию соединительной ткани, врожденные пороки развития сердечно-сосудистой системы (ДАК, КоА) и повышенную травматичность сосудистой стенки.
К структурным порокам аорты может приводить аномальный состав внеклеточного матрикса. Повышенная экспрессия матриксных металлопротеиназ и пониженная экспрессия тканевых ингибиторов матриксных металлопротеиназ (TIMP1 и TIMP3), выявляемые у пациенток с сШ-Т, могут привести к деградации компонентов внеклеточного матрикса стенки аорты, что приводит к ее истончению и увеличению диаметра [16].
Согласно литературным данным, распространенность сосудистых осложнений в данной когорте достигает 30% [17]. РА представляет собой жизнеугрожающее состояние, типично проявляющееся острой болью в груди, шее и/или спине и нередко осложняющееся проксимальным разрывом, гемоперикардом и тампонадой сердца. При сШ-Т РА обычно имеет фатальный исход, причем у молодых женщин с болевым синдромом этот диагноз зачастую не рассматривается.
При синдроме сШ-Т определен ряд ключевых факторов риска развития РА. Эпидемиологические исследования свидетельствуют о значительном снижении возраста манифестации РА у данной категории пациентов: медиана возраста при развитии РА или разрыва аорты составляет 30-35 лет, при этом до 24% случаев регистрируются у лиц моложе 20 лет, особенно на фоне сопутствующей аортопатии или КоА [18-20]. Вторым значимым фактором является кариотип и фенотип: наибольшая частота сердечно-сосудистых аномалий развития наблюдается у пациенток с кариотипом 45,X [21][22]. Третий фактор — АГ, распространенность которой при сШ-Т достигает приблизительно 50% [23]. Четвертый фактор — беременность, ассоциированная с высоким риском материнских осложнений, включая эклампсию; до 2% летальных исходов у беременных с сШ-Т обусловлены именно РА или разрывом аорты [24][25].
В представленном нами случае РА развилось на фоне эпизодической АГ, что подтверждает повышенную уязвимость сосудистой стенки при сШ-Т даже при транзиторном повышении АД. Клиническая картина отличалась атипичностью: доминировали неспецифические симптомы, что симулировало проявления нейроциркуляторной дистонии и психогенного расстройства. Подобные диагностические трудности признаны значимым фактором отсроченной диагностики РА [26]. Особый патогенетический интерес представляет назначение заместительной терапии L-тироксином незадолго до манифестации РА. Учитывая, что тиреоидные гормоны обладают положительным хроно- и инотропным эффектом, повышают чувствительность рецепторов к катехоламинам и потенцируют симпатическую активность, можно предположить, что даже кратковременная коррекция гипотиреоза на фоне сосудистой дисплазии и врожденных аномалий аорты могла индуцировать гипертензивную реакцию. Последняя, в свою очередь, могла выступить триггерным фактором РА [27-30]. Предполагаемый механизм тироксин-опосредованной гемодинамической нагрузки на морфологически измененное сосудистое русло требует дальнейшего изучения, но имеет патофизиологическое обоснование.
Сочетанная патология в виде сШ-Т, РА и ишемического инфаркта в правой половине подковообразной почки в данном наблюдении может отражать системный характер сосудистых нарушений, присущих данной хромосомной аномалии. Клинические и анатомические исследования подтверждают высокую частоту аномалий ветвления почечных артерий, их стенозов и инфарктов паренхимы почек при сШ-Т [16]. Ключевым методом верификации диагноза РА стала КТ-ангиография, остающаяся "золотым стандартом" диагностики [31][32].
D-димер (>0,5 мг/л) может быть использован в качестве индикатора типа РА и предиктора в оценке прогноза. Однако широкий спектр состояний, вызывающих его повышение (эмболия легочной артерии, коронарный тромбоз и др.), обуславливает его низкую специфичность [33][34].
Представленный клинический случай РА у молодой пациентки с сШ-Т подчеркивает судьбоносную необходимость поддержания высокой степени клинической настороженности среди врачей различных специальностей при ведении пациентов с врожденными генетическими патологиями соединительной ткани. Данная ситуация демонстрирует, что ключевым аспектом является не только понимание специфических рисков, ассоциированных с генетическим синдромом, но и тщательная оценка потенциальных триггерных факторов, способных инициировать острое сосудистое событие, к числу которых в описанном случае относится начало заместительной терапии тиреоидными гормонами.
Сложность и редкость подобных случаев обусловливают важность персонализированного подхода к диагностике и лечению, требующего интегрированного анализа генетической предрасположенности, клинической симптоматики и данных методов визуализации. Это наблюдение служит важным напоминанием о необходимости учета фонового состояния пациента и аргументирует потребность в разработке более точных клинико-диагностических алгоритмов для выявления васкулопатий у лиц с синдромами соединительнотканной дисплазии. Следовательно, представленный случай акцентирует значимость раннего выявления и динамического мониторинга патологии аорты у пациенток с сШ-Т, особенно при наличии эпизодической АГ, атипичного болевого синдрома и характерных фенотипических признаков.
Комплексный мультидисциплинарный подход, вклю-
чавший своевременное проведение компьютерно-томо-
графической ангиографии аорты и интерпретацию соответствующих лабораторных показателей, обеспечил корректную диагностику и направление пациентки в профильное кардиохирургическое отделение. Однако отсроченная диагностика, связанная с особенностями фенотипа пациентки, предопределила неблагоприятный летальный исход. На основании вышеизложенного мы рекомендуем внедрение регулярного скрининга диаметра аорты с использованием методов лучевой диагностики (предпочтительно МР-ангиографии, при ее недоступности — КТ-ангиографии), достижение строгого контроля АД и повышение осведомленности клиницистов о риске острых сосудистых событий у данной категории пациентов для оптимизации своевременной диагностики и профилактики жизнеугрожающих осложнений.
1. Khan N, Farooqui A, Ishrat R. Turner Syndrome where are we? Orphanet J Rare Dis. 2024;19(1):314. doi:10.1186/s13023-024-03337-0.
2. Gravholt CH, Viuff M, Just J, et al. The Changing Face of Turner Syndrome. Endocr Rev. 2023;44(1):33-69. doi:10.1210/endrev/bnac016.
3. Thunström S, Krantz E, Thunström E, et al. Incidence of Aortic Dissection in Turner Syndrome. Circulation. 2019;139(24):2802-4. doi:10.1161/CIRCULATIONAHA.119.040552.
4. Thunström S, Thunström E, Naessén S, et al. Aortic size predicts aortic dissection in Turner syndrome — A 25-year prospective cohort study. Int J Cardiol. 2023;373:47-54. doi:10.1016/j.ijcard.2022.11.023.
5. Thunström S, Thunström E, Naessén S, et al. All-cause mortality and death by aortic dissection in women with Turner syndrome: A national clinical cohort study. Am Heart J. 2025;281:1-9. doi:10.1016/j.ahj.2024.11.007.
6. Olukorode JO, Onwuzo CN, Otabor EO, et al. Aortic Size Index Versus Aortic Diameter in the Prediction of Rupture in Women With Abdominal Aortic Aneurysm. Cureus. 2024;16(4): e58673. doi:10.7759/cureus.58673.
7. Girardi LN, Lau C, Gambardella I. Aortic dimensions as predictors of adverse events. J Thorac Cardiovasc Surg. 2021;161(4):1193-7. doi:10.1016/j.jtcvs.2020.06.137.
8. Nijs J, Gelsomino S, Lucà F, et al. Unreliability of aortic size index to predict risk of aortic dissection in a patient with Turner syndrome. World J Cardiol. 2014;6(5):349-52. doi:10.4330/wjc.v6.i5.349.
9. Gravholt CH, Viuff MH, Brun S, et al. Turner syndrome: mechanisms and management. Nat Rev Endocrinol. 2019;15(10):601-14. doi:10.1038/s41574-019-0224-4.
10. Thuijs DJFM, Davierwala P, Milojeciv M, et al. Long-term survival after coronary bypass surgery with multiple versus single arterial grafts. European Journal of Cardio-Thoracic Surgery. 2022;61(4):925-33. doi:10.1093/ejcts/ezab392.
11. Isselbacher EM, Preventza O, Black JH, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association. American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022; e223-e393. doi:10.1161/CIR.0000000000001106.
12. Lombardi JV, Hughes GC, Appoo JJ, et al. Society for Vascular Surgery (SVS) and Society of Thoracic Surgeons (STS) reporting standards for type B aortic dissections. The Annals of thoracic surgery. 2020;109(3):959-81. doi:10.1016/j.athoracsur.2019.10.005.
13. Petrov I, Nedevska M, Chilingirova N, et al. Endovascular repair of dissecting thoracic aortic aneurysm in a patient with Turner syndrome. J Endovasc Ther. 2006;13(5):693-6. doi:10.1583/05-1663.1.
14. Blunden CE, Urbina EM, Lawson SA, et al. Progression of Vasculopathy in Young Individuals with Turner Syndrome. Pediatr Cardiol. 2021;42(3):481-91. doi:10.1007/s00246-020-02505-w.
15. Nóbrega PR, da Costa FBS, Rodrigues PGB, et al. Moyamoya associated with Turner syndrome in a patient with type 2 spinocerebellar ataxia-Occam’s razor or Hickam’s dictum: a case report. BMC Neurol. 2022;22(1):381. doi:10.1186/s12883-022-02912-x.
16. Yoon SH, Kim GY, Choi GT, et al. Organ Abnormalities Caused by Turner Syndrome. Cells. 2023;12(10):1365. doi:10.3390/cells12101365.
17. Milewicz DM, Braverman AC, De Backer J, et al. Marfan syndrome. Nat Rev Dis Primers. 2021;7(1):64. doi:10.1038/s41572-021-00298-7. Erratum in: Nat Rev Dis Primers. 2022;8(1):3. doi:10.1038/s41572-022-00338-w.
18. Meccanici F, de Bruijn JWC, Dommisse JS, et al. Prevalence and development of aortic dilation and dissection in women with Turner syndrome: a systematic review and meta-analysis. Expert Review of Cardiovascular Therapy. 2023;21(2):133-44. doi:10.1080/14779072.2023.2172403.
19. Bradley-Watson J, Glatzel H, Turner HE, et al. Elective Aortic Surgery for Prevention of Aortic Dissection in Turner Syndrome: The Potential Impact of Updated European Society of Cardiology and International Turner Syndrome Consensus Group Guidelines on Referrals to the Heart Team. Clin Endocrinol (Oxf). 2025;102(5):559-64. doi:10.1111/cen.15199.
20. Calanchini M, Bradley-Watson J, McMillan F, et al. Risk assessment for aortic dissection in Turner syndrome: The role of the aortic growth rate. Clin Endocrinol (Oxf). 2024;100(3):269-76. doi:10.1111/cen.15017.
21. Wu HH, Li H. Karyotype classification, clinical manifestations and outcome in 124 Turner syndrome patients in China. Ann Endocrinol (Paris). 2019;80(1):10-5. doi:10.1016/j.ando.2017.10.011.
22. Fiot E, Alauze B, Donadille B, et al. Turner syndrome: French National Diagnosis and Care Protocol (NDCP; National Diagnosis and Care Protocol). Orphanet J Rare Dis. 2022;17(1):261. doi:10.1186/s13023-022-02423-5.
23. Jones L, Blair J, Hawcutt DB, et al. Hypertension in Turner syndrome: a review of proposed mechanisms, management and new directions. J Hypertens. 2023;41(2):203-11. doi:10.1097/HJH.0000000000003321.
24. Whigham CA, Vollenhoven B, Vincent AJ. Reproductive health in Turner syndrome: A narrative review. Prenat Diagn. 2023;43(2):261-71. doi:10.1002/pd.6261.
25. Porcu E, Cipriani L, Damiano G. Reproductive health in Turner’s syndrome: from puberty to pregnancy. Front Endocrinol (Lausanne). 2023;14:1269009. doi:10.3389/fendo.2023.1269009.
26. Altenburg MM, Davis AM, DeCara JM. Diagnosis and Management of Aortic Diseases. JAMA. 2024;331(4):352-3. doi:10.1001/jama.2023.23668.
27. Evron JM, Hummel SL, Reyes-Gastelum D, et al. Association of Thyroid Hormone Treatment Intensity With Cardiovascular Mortality Among US Veterans. JAMA Netw Open. 2022;5(5):e2211863. doi:10.1001/jamanetworkopen.2022.11863.
28. Berta E, Lengyel I, Halmi S, et al. Hypertension in Thyroid Disorders. Front Endocrinol (Lausanne). 2019;10:482. doi:10.3389/fendo.2019.00482.
29. Lacka K, Pempera N, Główka A, et al. Turner Syndrome and the Thyroid Function-A Systematic and Critical Review. Int J Mol Sci. 2024;25(23):12937. doi:10.3390/ijms252312937.
30. Song Y, Yang H, Wang L, et al. Association of thyroid autoimmunity and the response to recombinant human growth hormone in Turner syndrome. J Pediatr Endocrinol Metab. 2021;34(4):465-71. doi:10.1515/jpem-2020-0610.
31. Spangenberg A, Rao SJ, Mackrell J, et al. Type A Aortic Dissection and Non-Contrast Computed Tomography. J Community Hosp Intern Med Perspect. 2023;13(3):118-20. doi:10.55729/2000-9666.1178.
32. Tekinhatun M, Akbudak İ, Özbek M, et al. Comparison of coronary CT angiography and invasive coronary angiography results. Ir J Med Sci. 2024;193(5):2239-48. doi:10.1007/s11845-024-03745-y.
33. Charnaia MA, Dement’eva II. Hemostasis system in the abdominal aorta aneurysms. Russian Journal of Cardiology and Cardiovascular Surgery. 2017;10(4):4-7. doi:10.17116/kardio20171044-7.
34. Carter JM, Tom RB, Sunesra R, et al. D-dimer as a Rule-Out for Aortic Dissection. Cureus. 2023;15(12):e50170. doi:10.7759/cureus.50170.
Врач-стажёр.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Алекберов Р. И. — Врач-кардиолог.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Врач-стажёр.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Директор.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Врач-эндокринолог.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Рустам Сабирович Талыбов — Зам. главного врача по развитию науки и инновациям.
Тюмень
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Бутенко Д.С., Алекберов Р.И., Спасенников В.В., Клещевникова Т.М., Талыбова А.Ю., Талыбов Р.С. Расслоение аорты у пациента с синдромом Шерешевского-Тернера. Клинический случай. . 2025;30(10S):6425. https://doi.org/10.15829/1560-4071-2025-6425. EDN: OUXIDK
Butenko D.S., Alekberov R.I., Spasennikov V.V., Kleshchevnikova T.M., Talybova A.Yu., Talybov R.S. Aortic dissection in a patient with Turner syndrome: a case report. . 2025;30(10S):6425. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6425. EDN: OUXIDK




