



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2025-6266
EDN: GXOHQC
Перейти к:
Введение. В статье представлено клиническое наблюдение за ребенком с синдромом удлиненного интервала QT и особенностями строения скелета, лицевыми аномалиями и умственной отсталостью. Этот симптомокомплекс явился проявлением ранее не описанного в России заболевания, наследуемого Х-сцеплено — синдрома Огдена, связанного с мутацией в гене NAA10.
Краткое описание. Молекулярно-генетическое обследование подтвердило мутацию (p.Tyr43Ser) в этом гене, которая также обнаружена у родной сестры и матери ребенка. В процессе наблюдения, во время проведения наркоза у мальчика произошла остановка сердца, обусловленная резкой брадикардией и асистолией, в связи с чем пациенту имплантирован кардиовертер-дефибриллятор.
Дискуссия. Синдром Огдена — заболевание, ассоциированное с мутациями в гене NAA10, которое может оставаться нераспознанным. Выраженные психоневрологические симптомы могут длительное время маскировать кардиологические проявления заболевания, включая синдром удлиненного интервала QT, нарушения ритма и проводимости, которые могут проявляться опасными жизнеугрожающими аритмиями и внезапной смертью.
Комолятова В.Н., Дмитриева А.В., Заклязьминская Е.В., Исланов И.О., Макаров Л.М. Синдром Огдена — ультраредкий вариант заболевания с риском внезапной смерти (первое описание в России). Клинический случай. Российский кардиологический журнал. 2025;30(10S):6266. https://doi.org/10.15829/1560-4071-2025-6266. EDN: GXOHQC
Komolyatova V.N., Dmitrieva A.V., Zaklyazminskaya E.V., Islanov I.O., Makarov L.M. Ogden syndrome — ultra-rare disease variant with a sudden death risk (first description in Russia): a case report. Russian Journal of Cardiology. 2025;30(10S):6266. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6266. EDN: GXOHQC
Синдром удлиненного интервала QT (СУИQT) — одна из самых частых каналопатий, встречающихся в детском возрасте, диагностика которого строится на критериях P. Schwartz, основным диагностическим критерием является продолжительность интервала QT [1]. Согласно представлениям о генетике СУИQT от 75% до 95% выявленных мутаций сосредоточены в 3 основных генах KCNQ1, KCNH2 и SCN5A [2][3]. Длительно занимаясь этой проблемой у детей, мы впервые столкнулись со случаем сочетанного течения множественных пороков развития опорно-двигательной системы, мозга и СУИQT, который сопровождался нарушением проводимости на электрокардиограмме (ЭКГ) и остановкой сердца с последующей успешной реанимацией.
Мальчик 9 лет, от женщины, страдающей алкогольной и наркотической зависимостью. Роды срочные, самостоятельные на 40 неделе. С рождения наблюдается неврологом по поводу задержки моторного и психоречевого развития, умственной отсталости; ортопедом по поводу сколиоза, деформации стоп. С 4-х лет — кратковременные приступы потери сознания, провоцируемые стрессом и физической нагрузкой, купирующиеся самостоятельно, расценивались как эпилептические припадки. Периодически получал антиконвульсанты, без эффекта. Случаи внезапной смерти в семье отрицают.
В декабре 2020г при магнитно-резонансной томографии тазобедренных суставов пережил остановку сердца, после успешной реанимации обратили внимание на удлинение интервала QT на ЭКГ. Ребенок направлен на консультацию в Центр синкопальных состояний и сердечных аритмий ФНКЦ детей и подростков ФМБА России.
При осмотре: физическое развитие очень низкое, дисгармоничное с дефицитом массы (рост 124 см, вес 19 кг), задержка психического развития. Множественные стигмы дизэмбриогенеза (микроцефалия, низко посаженные уши, широкая и плоская переносица, гипертелоризм, широкие передние зубы с большими промежутками, неполная синдактилия 3 и 4 пальцев; сколиоз), множественный кариес (рис. 1). Тоны сердца четкие, ритмичные, частота сердечных сокращений (ЧСС) 95 уд./мин, шумов нет. Живот мягкий, безболезненный, печень по краю реберной дуги.
На ЭКГ покоя — резкое отклонение электрической оси сердца (ЭОС) влево (угол α=-30), синусовый ритм, ЧСС 94 уд./мин. PQ — 140 мсек. QRS — 90 мсек, QT — 410 мсек, QTc — 522 мсек, умеренная тахикардия, блокада передней ветви левой ножки, удли-
нение интервала QT (рис. 2).
При холтеровском мониторировании — синусовый ритм, умеренная тахикардия, редкие (<1%) одиночные и парные мономорфные желудочковые экстрасистолы, удлинение интервала QT (QT на минимальной ЧСС 49 уд./мин — 592 мсек, среднесуточный интервал QTс — 524 мсек). Зарегистрированы эпизоды альтернации зубца Т (изменение полярности Т волны в следующих друг за другом сокращениях) — грозный предвестник опасных желудочковых аритмий (рис. 3). По результатам эхокардиографии размеры сердца и сократительная способность в норме.
Молекулярно-генетическое исследование выявило миссенс мутацию p.Tyr43Ser в гене NAA10 (вероятно, патогенная, класс IV) в гемизиготном состоянии, которая ранее была описана у пациентов с синдромом Огдена. Других патологических мутаций у пациента не выявлено. Каскадный семейный скрининг выявил носительство этой мутации у матери и клинически здоровой сестры 18 лет, подтверждено капиллярным секвенированием по Сенгеру (рис. 4).
Клинический диагноз
Синдром Огдена: СУИQT, синкопальная форма, редкая желудочковая экстрасистолия, блокада передней ветви левой ножки пучка Гиса, задержка физического и психического развития, S-образный сколиоз грудного отдела позвоночника.
Динамика заболевания
На протяжении 3-х лет состояние ребенка оставалось стабильное, обмороков не было, получал бета-адреноблокаторы в дозе 2 мг/кг. Через 3 года, при лечении кариеса под общей анестезией, развилась критическая брадикардия и остановка сердца, потребовавшая реанимационных мероприятий. После восстановления сердечной деятельности кроме удлинения интервала QT на ЭКГ появилась атриовентрикулярная блокада 1 степени и транзиторная полная блокада левой ножки пучка Гиса (рис. 5).
После перенесенной остановки сердца пациент направлен на постановку имплантируемого кардиовертера-дефибриллятора (ИКД), которая проведена ему в декабре 2024г.
В последнее время у ребенка появилась дилатация левого желудочка и снижение сократительной способности миокарда (фракция выброса 43-44%), в крови повысился уровень тропонина I и N-концевого промозгового натрийуретического пептида, в терапию добавлены ингибиторы ангиотензинпревращающего фермента и спиронолактон.
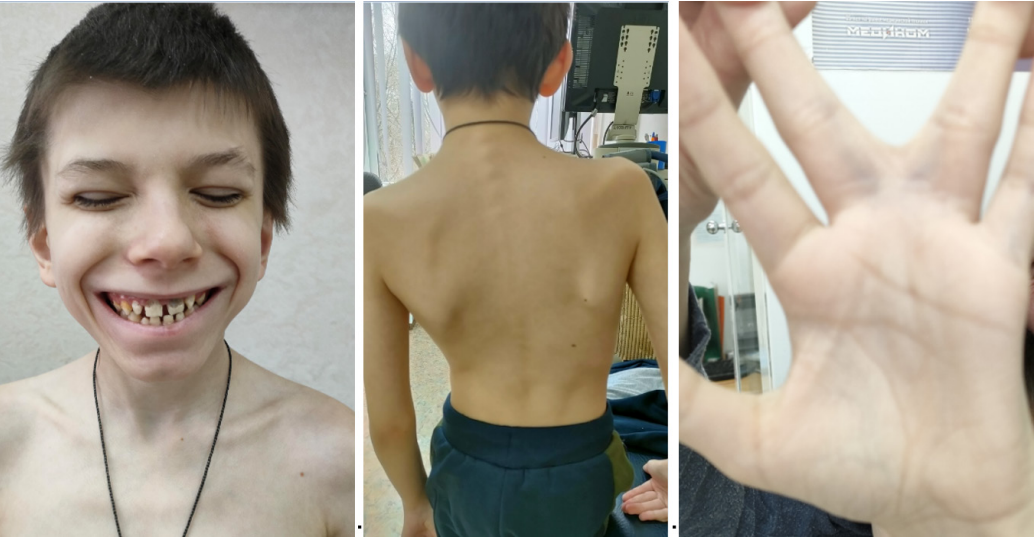
Рис 1. Фото пациента 9 лет с редким вариантом СУИQT.
Примечание: множественные стигмы дисэмбриогенеза (микроцефалия, низко посаженные уши, широкая и плоская переносица, гипертелоризм, широкие передние зубы, с большими промежутками, неполная синдактилия 3 и 4 пальцев; сколиоз). Фотографии демонстрируются с разрешения отца.
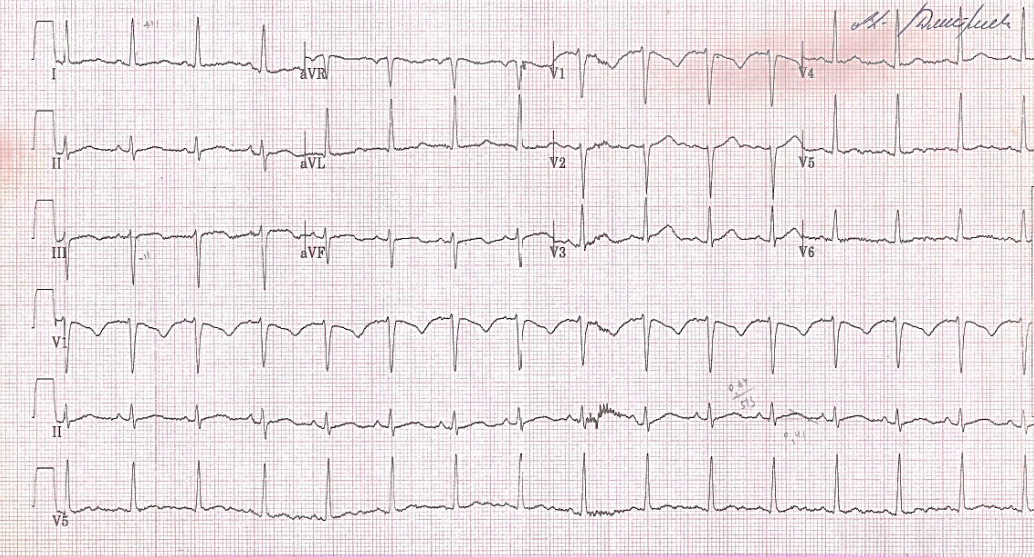
Рис. 2. ЭКГ покоя мальчика 9 лет с редким вариантом СУИQT.
Примечание: резкое отклонение ЭОС влево (угол α=-30), синусовый ритм, ЧСС 94 уд./мин. PQ — 140 мсек. QRS — 90 мсек, QT — 410 мсек, QTc — 522 мсек. Умеренная тахикардия, блокада передней ветви левой ножки пучка Гиса, удлинение интервала QT.
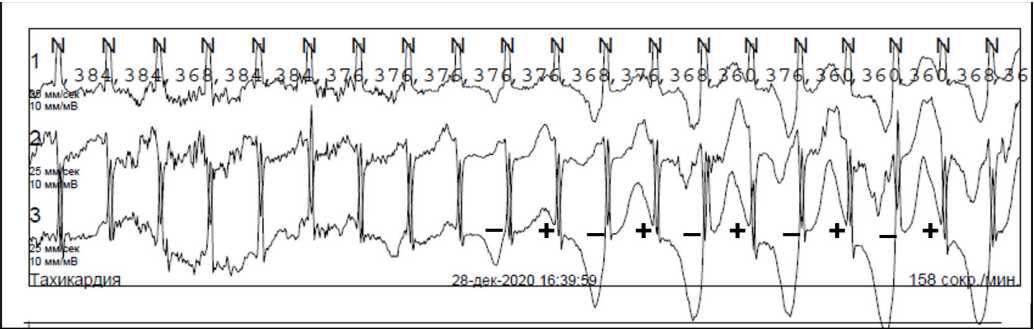
Рис. 3. Макроальтернации зубца Т при холтеровском мониторировании у ребенка 9 лет с редким вариантом СУИQT. Последовательное изменение полярности зубца Т (от высокого положительного до глубокого отрицательного).
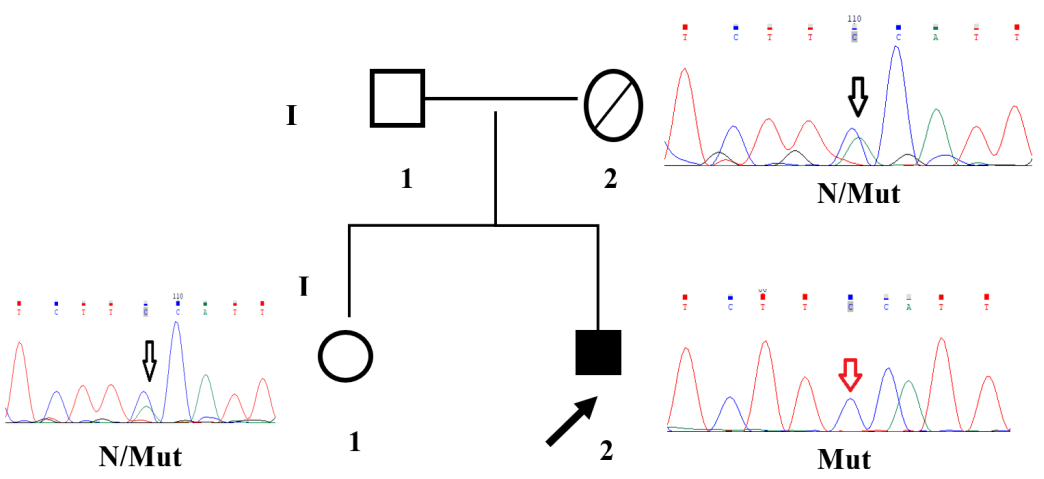
Рис. 4. Родословная семьи.
Примечание: здоровые члены семьи обозначены незакрашенными фигурами, пробанд с диагнозом обозначен сплошным заполнением. Через N обозначена референсная последовательность, Mut — замена NAA10 NM_003491.4: c.128A>C (p.Tyr43Ser). Через косую черту обозначено гетерозиготное состояние, пробанд — гемизиготное состояние.
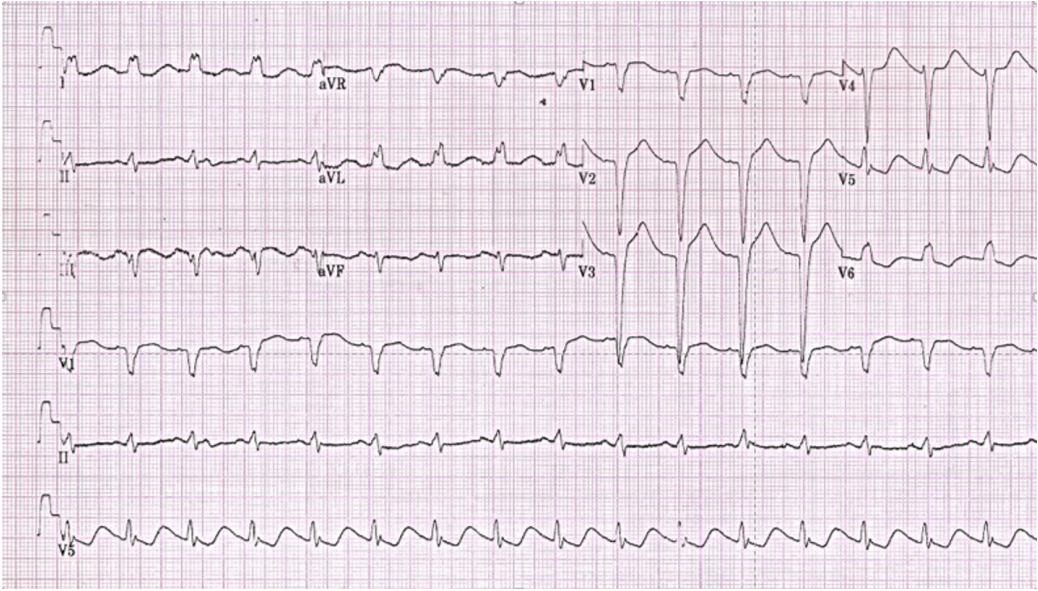
Рис. 5. ЭКГ мальчика 12 лет после восстановления сердечной деятельности.
Примечание: резкое отклонение ЭОС влево (угол α=-30). Синусовый ритм, тахикардия, ЧСС 102 уд./мин. PQ — 200 мсек. QRS — 135 мсек, QT — 420 мсек, QTc — 542 мсек, QTс (Багасян) — 533 мсек [4]. Умеренная тахикардия, полная блокада левой ножки пучка Гиса, атриовентрикулярная блокада 1 степени, удлинение интервала QT.
Представленный нами клинический случай интересен сочетанием СУИQT с различными нарушениями психического развития и патологией опорно-двигательной системы. Занимаясь много лет проблемой СУИQT, мы не встречались ранее с такими больными. Мы предполагали, что эти изменения существуют у пациента в контексте нескольких диагнозов, т.к. особенности его анамнеза позволяли предполагать наличие фетального алкогольного синдрома, который имеет схожий симптомокомплекс (грубое лицо, нарушение строения зубов, врожденные пороки развития, включая врожденные пороки сердца и скелетные аномалии) [5]. Но выявленная мутация в гене NAA10 позволила нам предположить крайне редкий, ранее не описанный в России, фенотип СУИQT — синдром Огдена.
Синдром Огдена, также известный как синдром нарушения развития нервной системы, является редким генетическим заболеванием, связанным с патологией в гене NAA10, генетические поломки приводят к дефициту N-терминальной ацетилтрансферазы. Синдром характеризуется целым рядом неврологических симптомов, включая умственную отсталость и судороги, задержку психического развития, врожденные пороки сердца, гипотонию и др. Синдром Огдена был назван в честь родного города первой семьи, наблюдаемой John M Opitz, с Х-сцепленным рецессивным заболеванием, характеризующимся ранней летальностью из-за структурных аномалий сердца и/или аритмий, тяжелой задержкой развития, постнатальной задержкой роста, прогерией с уменьшенной подкожной жировой тканью и избыточной кожей [6, 7].
Спектр клинических проявлений синдрома очень широкий, начиная от тяжелого фенотипа у мужчин с мутациями p.Ser37Pro в гене NAA10, первоначально описанного как синдром Огдена, до более легкой умственной отсталости, обнаруживаемой как у мужчин, так и у женщин. Задержка развития, интеллектуальные расстройства являются характерными признаками болезни (а в некоторых случаях и единственным проявлением), однако у многих пациентов выявляются и различные сердечно-сосудистые проявления [7]. В одном из недавно опубликованных обзоров отмечается, что нарушения ритма сердца у таких больных могут проявляться как внутриутробно, так и манифестировать после рождения [8]. Их спектр — обширный, как тахи-, так и брадиаритмии: суправентрикулярные или желудочковые экстрасистолы, тахикардия "torsades de pointes", атриовентрикулярная блокада 1 степени, удлинение интервала QT и др. Кроме нарушений ритма могут быть врожденные пороки сердца: открытый артериальный проток, дефекты межжелудочковой и межпредсердной перегородок, стеноз легочной артерии и др. Эти описания позволяют предположить, что выявленные изменения у нашего пациента существуют в рамках единого заболевания NAA10 синдрома или синдрома Огдена.
В литературе нам встретилось лишь одно наблюдение за семьей, где у двоих братьев и их матери была выявлена схожая мутация (p.Tyr43Ser) в гене NAA10 [9]. На фотографиях двух братьев в этом исследовании те же лицевые аномалии, что и у нашего пациента. У обоих сыновей и у их матери наблюдалось выраженное удлинение интервала QT (QTc >500 мсек). Одному из братьев и его матери после остановки сердца имплантирован ИКД. У другого брата выявлена гипертрофическая кардиомиопатия.
Генез сердечной недостаточности, наблюдаемой у нашего пациента, остается неясен, ее первые проявления выявлены после перенесенной вирусной инфекции и могли стать проявлением активного воспалительного процесса в миокарде. В связи с чем было бы целесообразно проведение магнитно-резонансной томографии сердца, однако выполнение его требует анестезии, что было невозможно в связи с остановкой сердца на фоне наркоза. Мы надеемся, что это станет реально после имплантации ИКД.
В одном из самых крупных обзоров по оценке фенотипических проявлений у больных с NAA10 синдромом показано, что в 17,9% случаев имелось удлинение интервала QTс, в таком же количестве — дефект межпредсердной перегородки, у 13,7% — дефект межжелудочковой перегородки, у 9,5% — гипертрофическая кардиомиопатия и различные нарушения ритма, у 6,3% — случаи остановки сердца [10]. В этой работе, как и в большинстве других, обсуждается возможная остановка сердца у таких пациентов по причине тахикардии типа "torsades de pointes". Однако в нашем случае зафиксированная остановка сердца произошла вследствие асистолии, что не типично не только для таких больных, но и в целом для пациентов с СУИQT, генез данного состояния остается для нас не ясен. Возможная причина — сочетание СУИQT и прогрессирующего поражения проводящей системы сердца, т.к. блокада передней ветви левой ножки пучка Гиса регистрировалась уже на первой ЭКГ. В литературе нам не встретилось описаний нарушений проводимости у больных с мутациями в гене NAA10, хотя в некоторых обзорах упоминается о возможных брадиаритмиях у таких пациентов [8].
Синдром Огдена — заболевание, ассоциированное с мутациями в гене NAA10, является редким мультисистемным заболеванием, которое может оставаться нераспознанным. Выраженные психоневрологические симптомы могут длительное время маскировать кардиологические проявления заболевания, которые включают СУИQT, нарушения ритма и проводимости, проявляющиеся опасными жизнеугрожающими аритмиями и внезапной смерти.
1. Schwartz P, Moss A, Vincent G, et al. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-4. doi:10.1161/01.cir.88.2.782.
2. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308-39. doi:10.1016/j.hrthm.2011.05.020.
3. Adler A, Sadek MM, Chan AY, et al. Patient outcomes from a specialized inherited arrhythmia clinic. Circ Arrhythm electrophysiol. 2016;9:e003440. doi:10.1161/CIRCEP.115.003440.
4. Комолятова В. Н., Шаблинова Т. С., Дроздов Д. В. и др. Интервал QT на электрокардиограмме покоя: значение и методы измерения. Вестник аритмологии. 2024;31(2):е15-е23. doi:10.35336/VA-1301.
5. Lind JN, Interrante JD, Ailes EC, et al. Maternal Use of Opioids During Pregnancy and Congenital Malformations: A Systematic Review. Pediatrics. 2017;139(6):e20164131. doi:10.1542/peds.2016-4131.
6. Rope AF, Wang K, Evjenth R, et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants dueto N-terminal acetyltransferase deficiency. American Journal of Human Genetics. 2011;89(1):28-43. doi:10.1016/j.ajhg.2011.05.017.
7. Gogoll L, Steindl K, Joset P, et al. Confirmation of Ogden syndrome as an X-linked recessive fatal disorder due to a recurrent NAA10 variant and review of the literature. J Med Genet. A.2021;185(8):2546-60. doi:10.1002/ajmg.a.62351.
8. Wu Y, Lyon GJ. NAA10-related syndrome. Experimental & Molecular Medicine. 2018;50(7):1-10. doi:10.1038/s12276-018-0098-x.
9. Casey JP, Støve SI, McGorrian C, et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Sci Rep. 2015;2(5):16022. doi:10.1038/srep16022.
10. Lyon GJ, Vedaie M, Beisheim T, et al. Expanding the phenotypic spectrum of NAA10-related neurodevelopmental syndrome and NAA15-related neurodevelopmental syndrome. European Journal of Human Genetics. 2023;31:824-33. doi:10.1038/s41431-023-01368-y.
Вера Николаевна Комолятова — д.м.н., врач Центра синкопальных состояний и сердечных аритмий, профессор кафедры педиатрии им. Н. Г. Сперанского.
Москва
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Алёна Владимировна Дмитриева — врач Центра синкопальных состояний и сердечных аритмий.
Москва
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Елена Валерьевна Заклязьминская — зав. лабораторией медицинской генетики, профессор кафедры медицинской генетики.
Москва
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Игорь Олегович Исланов — лаборатория медицинской генетики.
Москва
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Леонид Михайлович Макаров — д.м.н., профессор, руководитель Центра синкопальных состояний и сердечных аритмий, профессор кафедры педиатрии им. Н. Г. Сперанского.
Москва
все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье
Комолятова В.Н., Дмитриева А.В., Заклязьминская Е.В., Исланов И.О., Макаров Л.М. Синдром Огдена — ультраредкий вариант заболевания с риском внезапной смерти (первое описание в России). Клинический случай. Российский кардиологический журнал. 2025;30(10S):6266. https://doi.org/10.15829/1560-4071-2025-6266. EDN: GXOHQC
Komolyatova V.N., Dmitrieva A.V., Zaklyazminskaya E.V., Islanov I.O., Makarov L.M. Ogden syndrome — ultra-rare disease variant with a sudden death risk (first description in Russia): a case report. Russian Journal of Cardiology. 2025;30(10S):6266. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6266. EDN: GXOHQC













































