



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2024-6016
Перейти к:
Современные исследования демонстрируют, что клональный гемопоэз неопределенного потенциала (КГНП) является фактором риска развития и прогноза хронической сердечной недостаточности (ХСН) различной этиологии. Патофизиология и последствия КГНП носят геноспецифичный характер. Механизмы, задействованные в этом процессе, сложны и указывают на центральную роль системного и миокардиального воспаления, включая иммунный ответ, зависящий от каскада инфламмасома/интерлейкин-1β/интерлейкин-6. КГНП и ассоциируемые с ним воспалительные пути представляют собой мощную потенциальную мишень, что обусловливает актуальность научных работ в зоне различных стадий ХСН и маркеров этого генетического феномена. Лучшее понимание взаимодействий между мутантными клонами, иммунными путями, хроническим воспалением и клинической реализацией ХСН может иметь важное значение в контексте прецизионной и персонализированной медицины.
Лясникова Е.А., Иванченко Л.Ю., Козлова С.Н., Ситникова М.Ю., Костарева А.А., Шляхто Е.В. Клональный гемопоэз неопределённого потенциала и хроническая сердечная недостаточность. Российский кардиологический журнал. 2024;29(11S):6016. https://doi.org/10.15829/1560-4071-2024-6016
Lyasnikova E.A., Ivanchenko L.Yu., Kozlova S.N., SITNIKOVA M.Yu., Kostareva A.A., Shlyakhto E.V. Clonal hematopoiesis of indeterminate potential and heart failure. Russian Journal of Cardiology. 2024;29(11S):6016. (In Russ.) https://doi.org/10.15829/1560-4071-2024-6016
Растущая проблема хронической сердечной недостаточности (ХСН) по-прежнему ассоциирована с высокой инвалидизацией и смертностью, что определяет поиск мер профилактической направленности. Использование шкал прогноза в алгоритмах маршрутизации пациентов поддерживается современными рекомендациями и может оптимизировать лечение [1]. Вместе с тем прогнозирование исходов у больных с сердечной недостаточностью (СН) затруднено из-за постарения населения и мультиморбидности. Поиск новых риск-стратифицированных маркеров и усовершенствование методов прогноза у пациентов с ХСН в условиях постоянного улучшения подходов к менеджменту, разработки новых терапевтических возможностей и цифровой обработки информации продолжается.
Многочисленные исследования подтверждают связь системного воспаления как с факторами кардиометаболического риска, непосредственно атеросклерозом, так и СН. Являясь причинно-следственным звеном при СН, воспаление играет немаловажную роль в патогенезе заболевания и ассоциировано с её неблагоприятным исходом независимо от традиционных показателей, таких как фракция выброса (ФВ) левого желудочка (ЛЖ) и функциональный класс (ФК) [2][3]. Принимая во внимание этот факт, в последнее десятилетие активно ведется поиск мишеней, ассоциируемых с воспалением, для профилактических и лечебных вмешательств в зоне ХСН.
Недавние исследования в области технологии полногеномного секвенирования нового поколения позволили выявить клональный гемопоэз с неопределенным потенциалом (КГНП) (clonal hematopoiesis of indeterminate potential (CHIP)), характеризующийся приобретенными соматическими мутациями с лейкогенным потенциалом в течение жизни и формированием популяций мутантных клонов провоспалительных иммунных клеток, присутствующих в костном мозге и циркуляции. КГНП рассматривается как своеобразная мутационно-специфическая адаптация к изменению окружающей среды. Опубликованные данные указывают на то, что генетическая предрасположенность и внешние факторы, такие как химиотерапия и радиация, наряду с метаболическими и воспалительными стрессами способствуют соматическим мутациям гемопоэтических стволовых клеток, что приводит к преимуществам клеточного выживания и клональному расширению (экспансии) клеток этой линии [4]. КГНП на текущий момент определяется, если частота вариантного аллеля (Variant Allele Fraction (VAF)) мутации "драйверного" гена КГНП ≥2%, при этом критерии гематологической неоплазии, дисплазии или цитопении отсутствуют [5][6]. Надо отметить, что современные методы технологии секвенирования позволяют обнаруживать мутации КГНП с гораздо более низкой частотой. В любом случае это распространенное явление, тесно связанное со старением и способствующее формированию генетически отличной субпопуляции циркулирующих лейкоцитов, в последние 5 лет все чаще рассматривается как фактор риска (ФР) ряда возраст-ассоциированных состояний, включая не только гемобластозы, но и сердечно-сосудистые заболевания (ССЗ). Впервые Jaiswal S, et al. (2014) продемонстрировали связь КГНП с общей смертностью и двукратным увеличением риска развития атеросклеротических заболеваний, таких как ишемическая болезнь сердца и ишемический инсульт [7]. Этой же группой ученых у носителей мутаций КГНП были выявлены более выраженная кальцификация коронарных артерий по данным компьютерной томографии и более высокая частота развития инфаркта миокарда в молодом возрасте (у мужчин <40 лет и женщин <50 лет) по сравнению с лицами без КГНП [8]. Дальнейшие клинические и экспериментальные исследования на животных показали связь КГНП с большим спектром и других ССЗ, в патогенезе которых задействованы различные механизмы воспаления, включая аортальный стеноз, венозный тромбоз и легочную гипертензию [9-12]. В последнее время накапливаются данные, указывающие на роль КГНП в развитии и прогнозе СН, как ишемической, так и неишемической этиологии, чему и посвящён представленный обзор.
Надо подчеркнуть, что клональная экспансия клеток гемопоэтической системы с генетическими изменениями ("драйверными" мутациями), дающими этим клеткам определенные преимущества в пролиферации и/или устойчивость к неблагоприятным факторам по сравнению с остальными гемопоэтическими клетками, увеличивается с возрастом и обнаруживается в основном после 50 лет. В указанном возрастном диапазоне у большинства индивидуумов присутствуют бремя кардиометаболических рисков, проявления патологии сердечно-сосудистой системы той или иной степени выраженности, доклинические и клинически выраженные стадии СН, что может затруднять оценку причинно-следственных связей. Распространенность КГНП очень низка в возрасте до 40 лет, у лиц в возрасте 60 лет достигает примерно 5% и резко возрастает до 30-40% у людей старше 80 лет [7]. Однако ввиду непостоянства темпов накопления мутантных клонов признаки клонального гемопоэза могут наблюдаться и в более молодом возрасте [8][13-15].
Механизмы, ответственные за развитие КГНП и его связь с ССЗ, изучены лишь частично. К основным процессам, связанным с превалированием созревания одних клонов над другими при КГНП, относят: потерю баланса между самообновлением и дифференцировкой гемопоэтических стволовых клеток, повышение устойчивости клонов к внешним воздействиям и защиту клона от воспаления. Чаще всего "драйверные" мутации КГНП, ассоциируемые с ССЗ, описаны в генах, кодирующих эпигенетические регуляторы (DNMT3A, TET2 и ASXL1), сигнальные белки медиаторов воспаления (JAK2), компоненты сплайсинга (SRSF2 и SF3B1) и факторы, участвующие в процессах повреждения ДНК и клеточной гибели (PPM1D и TP53) [16][17]. Немаловажно подчеркнуть, что спектр и частота генетических вариантов, ассоциируемых с КГНП и сердечно-сосудистыми событиями, непрерывно обсуждаются и продолжают уточняться. В ряде единичных популяционных исследований не было продемонстрировано зависимости между наиболее частыми мутациями генов DNMT3A, TET2 и ASXL1 и коронарной болезнью сердца, а также ишемическим инсультом. В то же время КГНП наряду с возрастом и гематологическими неоплазиями, был связан с курением и наличием/дебютом коморбидностей, ассоциируемых с данной вредной привычкой: хронической обструктивной болезнью легких (ХОБЛ), эмфиземой, раком легкого, заболеваниями периферических артерий [18].
На текущий момент существуют убедительные доказательства, что негативные эффекты КГНП в развитие ССЗ носят геноспецифичный характер, но имеют общий знаменатель, а именно способствуют провоспалительному состоянию, которое характерно для стареющих тканей, из-за усиленного производства мутантными клетками ряда медиаторов воспаления, включая интерлейкин (ИЛ)-1 бета (ИЛ-1β) и ИЛ-6 [14][19]. Результаты исследований на животных моделях показывают, что серия мутаций, связанных с этим феноменом, формируют ответ мутантного клона на течение иммунологических процессов. Потомство мутантных клонов генерирует воспалительную среду, к которой оно устойчиво, это дает определённое преимущество перед клетками без мутаций, способствуя клональному доминированию [20][21].
В экспериментальных исследованиях обнаружена связь мутации TET2 с повышенной экспрессией моноцитами ИЛ-1β и ИЛ-6 непосредственно или опосредованно через NLRP3-инфламмасомный путь, в т. ч. в самом миокарде при СН [22][23]. C помощью РНК-секвенирования единичных клеток продемонстрирована повышенная экспрессия воспалительных генов каскада NLRP3-инфламмасома/ИЛ-1β/ИЛ-6 в моноцитах пациентов с СН, несущих мутации DNMT3A, по сравнению с моноцитами, полученными от пациентов с СН без мутаций данного гена. Более того, в этой работе носители мутации DNMT3A дополнительно характеризовались воспалительным транскриптомом, ассоциированным с увеличением экспрессии факторов, способствующих адгезии моноцитов к эндотелию и активации Т-клеточного звена иммунной системы [24]. Эксперименты с использованием методологии лентивирусного вектора/CRISPR/Cas9 выявили, что инактивация TET2 и DNMT3A одинаково способствовала индукции ангиотензин II опосредованной гипертрофии и дисфункции миокарда, сердечному и почечному фиброзу, имея, однако, геноспецифичные различия в экспансии мутантных гемопоэтических клеток в миокард и различные паттерны экспрессии провоспалительных молекул. Деактивация TET2 способствовала экспрессии лиганда 5 мотива хемокина CC (CСL5), тогда как при деактивации DNMT3A увеличивалась экспрессия лигандов CXC хемокинов макрофагами [25].
В экспериментальных мышиных моделях СН с низкой ФВ (СНнФВ) ишемической и неишемической этиологии с мутациями p.V617F в гене JAK2 КГНП продемонстрированы активация инфламмасомного комплекса через другой белок-сенсор — AIM2, с последующей повышенной экспрессией ИЛ-1 и ИЛ-6 макрофагами в миокарде мышей и более выраженные неблагоприятные структурно-функциональные изменения сердечной мышцы по сравнению с животными без мутаций [26][27]. На сегодняшний день многочисленные данные подтверждают, что при наличии мутации JAK2 значительно возрастают риски венозного и артериального тромбоза, в т. ч. вследствие увеличения образования нейтрофильных внеклеточных ловушек, способствующих дисрегуляции гемостаза, при таких провоспалительных состояниях, как рак и атеросклероз [10][28-30].
Помимо вышеперечисленных цитокинов, повышение фактора некроза опухоли альфа (ФНО-α) наблюдалось в циркулирующих моноцитах пациентов с СН (включая пациентов с аортальным стенозом) с DNTM3A и TET2 "драйверными" мутациями, а также в мышиных моделях СН с мутацией JAK2 [26][31].
Менее известно о том, как мутации ASXL1, SF3B1, SRSF2 и другие "драйверные" гены КГНП влияют на иммунную функцию. Продемонстрировано, что более высокий уровень циркулирующего ИЛ-6 наблюдался у носителей ASXL1, тогда как уровень циркулирующего ИЛ-18 повышался у лиц с SF3B1 [16].
Очевидно, мутации TP53 и PPM1D КГНП, ассоциируемые с повреждением ДНК, распространены после химио- или лучевой терапии онкологических заболеваний и могут быть связаны с различными типами миокардиальной дисфункции. На мышиных моделях было показано, что мутации TP53 приводят к росту антрациклиновой кардиотоксичности в т. ч. за счет усиленного транспорта нейтрофилов в поврежденный миокард и повышения экспрессии ИЛ-1β, ИЛ-6 и ФНО-α [32]. Аналогично вышеприведенным экспериментальным исследованиям по TЕТ2 мутации PPM1D, полученные с помощью CRISPR/Cas9 технологии, вызывали более высокие уровни экспрессии ИЛ-1β макрофагами, а неблагоприятные ремоделирующие эффекты на миокард при введении ангиотензина II были предотвращены с помощью ингибитора NLRP3 [33]. Этот факт позволяет предположить, что PPM1D мутации опосредуют кардиотоксичность и неблагоприятное ремоделирование сердца, по крайней мере, частично, через тот же цитокиновый путь, что и мутации TЕТ2 [12].
Важно отметить, что воспаление само по себе и провоспалительные цитокины, в частности, способны стимулировать клональную пролиферацию и экспансию гемопоэтических клеток мутировавших клонов [4][34][35]. Утяжеление провоспалительного статуса наряду с увеличением пула мутантных миелоидных клеток, ускоряет атеросклероз и ассоциированные с ним осложнения. Интерлейкины (ИЛ-1β, ИЛ-6, ИЛ-18), ФНО-α, другие воспалительные молекулы, непосредственно миокардиальное воспаление способствуют гипертрофии и фиброзу миокарда, и, как следствие, дезадаптивному ремоделированию сердца и СН [17][23][28][31][36][37]. Несмотря на то, что иммунологические пути, связанные с возникновением КГНП и риском ССЗ, продемонстрированные в эксперименте, требуют дальнейшего изучения в клинике, в целом, большое количество данных указывает на наличие петли положительной обратной связи между КГНП и воспалением, лежащем в основе ряда возраст-ассоциированных заболеваний сердечно-сосудистого континуума, исходом которого является СН [20][38] (рис. 1).
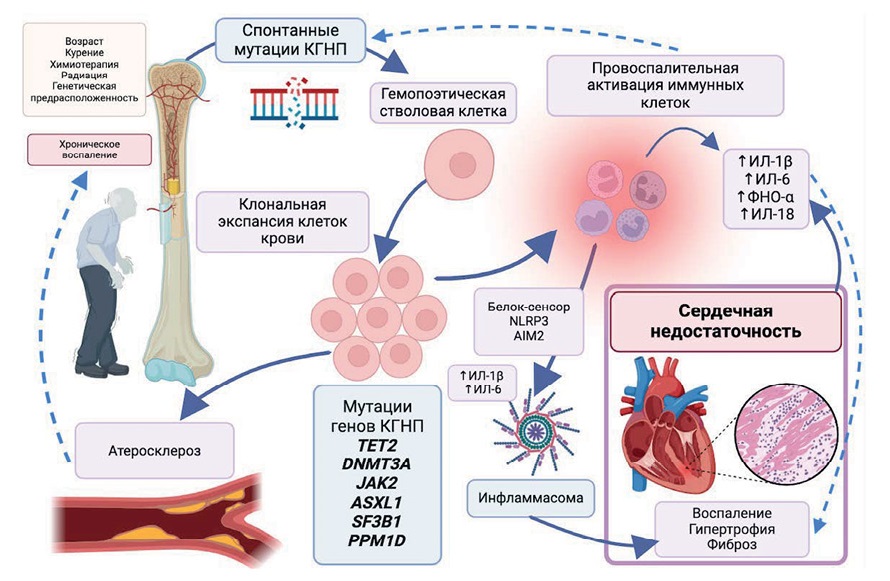
Рис. 1. КГНП, воспаление и СН.
Сокращения: ИЛ — интерлейкин, КГНП — клональный гемопоэз неопределенного потенциала, ФНО-α — фактор некроза опухоли.
Надо констатировать, что ФР или ассоциированные заболевания КГНП являются признанными классическими факторами, увеличивающими вероятность развития СН. Возраст, курение, малоподвижный образ жизни, нездоровая диета, терапия по поводу онкологического заболевания, преждевременная менопауза, хронические инфекции, аутоиммунные заболевания и загрязнения окружающей среды предрасполагают к соматическим мутациям и КГНП, являясь одновременно причинами развития патологий доклинических стадий ХСН [5][39].
КГНП глубоко взаимосвязан с дебютом и прогрессированием таких провоспалительных состояний, лежащих в основе возникновения СН преимущественно с сохраненной ФВ (СНсФВ), как ожирение, сахарный диабет (СД) 2 типа, ХОБЛ и хроническая болезнь почек. Ряд исследований связывают КГНП с инсулинорезистентностью и метаболическим синдромом [40]. Однако продемонстрировано, что и эти коморбидные состояния, в свою очередь, способны стимулировать клональный гемопоэз [4][17][41-43].
Представленные нами ранее доклинические данные о том, что мутации КГНП усиливают кардиотоксичность антрациклинов, подтверждены в исследовании на людях: у взрослых пациентов с лимфомой, получавших лечение доксорубицином, исходное наличие мутаций TET2 в 5 раз увеличивало риск снижения ФВ ЛЖ в течение 4,3 лет наблюдения [44].
Надо подчеркнуть, что исследования связи ФР ССЗ с КГНП все еще ограничены и не всегда имеют однозначный характер. Предполагается, что дислипидемия может усиливать провоспалительный эффект КГНП, в то же время клинические работы не подтвердили повышенную распространенность гиперхолестеринемии у пациентов с "драйверными" мутациями клонального гемопоэза [17]. Более того, курение, являясь признанным виновником риска ССЗ, значимо повышает вероятность данного феномена. Вместе с тем в экспериментальных исследованиях на животных показано, что соматические мутации связаны с развитием и тяжестью ХОБЛ, но не зависят от возраста и кумулятивного воздействия дыма [41].
Ряд клинических обсервационных работ последних 5 лет демонстрирует тесную связь между носительством мутаций КГНП и фактом возникновения, а также исходом СН. Спектр, встречаемость и прогностическая значимость генетических вариантов КГНП у пациентов с ХСН продолжают уточняться. Обзор текущих клинических данных по КГНП и СН представлен в таблице 1.
Таблица 1
Обзор текущих клинических данных по КГНП и СН
|
Автор, год |
Когорта, дизайн исследования |
Гены КГНП* |
Результаты исследования |
|
Yu B, et al. (2021) [45] |
56597 участников: 5 популяционных когорт (ARIC, CHS; JHS; UKBB; WHI) Средний возраст 58 лет 3406 (6%) случаев КГНП Наблюдение 20 лет: у 4694 (8,3%) развитие СН |
DNMT3A, TET2, ASXL1, JAK2 |
Увеличение риска развития СН (ОP 1,25; 95% ДИ: 1,13-1,38) при мутациях в генах ASXL1, TET2 и JAK2, но не DNMT3A Увеличение риска развития СН для больших клонов (VAF >10%): ОР 1,29; 95% ДИ: 1,15-1,44 |
|
Scolari FL, et al. (2022) [47] |
341 пациент с кардиогенным шоком (преимущественно неишемической этиологии) ФВ ЛЖ 26±12% vs 345 пациента амбулаторной выборки с СНнФВ ФВ ЛЖ 27±10% Медиана возраста для обеих групп 58 лет Наблюдение: 30 дней; 90 дней; 3 года |
DNMT3A, TET2, ASXL1 |
У пациентов с кардиогенным шоком наблюдается более высокая распространенность КГНП (ОШ 1,5; 95% ДИ: 1,0-2,1) Снижение выживаемости среди пациентов с КГНП в разных временных точках: 30 дней: ОР 2,7; 95% ДИ: 1,3-5,7; 90 дней: ОР 2,2; 95% ДИ: 1,3-3,9; 3 года: ОР 1,7; 95% ДИ: 1,1-2,8 |
|
Shi C, et al. (2023) [46] |
705 участников: Когорта PREVEND Медиана возраста 65 лет Наблюдение 11,5 лет Сравнение 358 пациентов с дебютом СНнФВ/СНсФВ vs 347 пациентов без СН |
DNMT3A, TET2, ASXL1 |
КГНП коррелировал с факторами риска и биомаркерами СН Увеличение риска развития СН во всей исследуемой популяции при мутации DNMT3A (ОР 1,52; 95% ДИ: 1,10-2,10) КГНП увеличивал риск развития СН у лиц <65 лет |
|
Автор, год |
Когорта, дизайн исследования |
Гены КГНП* |
Результаты исследования |
|
Dorsheimer L, et al. (2019) [48] |
200 пациентов с СНнФВ II ФК ишемической этиологии Когорта исследований эффектов интракоронарного введения аутологичных СКК Медиана возраста 65 лет Анализ мононуклеарных клеток костного мозга 38 (18,5%) случаев КГНП Наблюдение 4,4 года |
DNMT3A, TET2 |
Более высокая смертность с госпитализацией по поводу СН среди носителей DNMT3A/TET2 (ОР 2,1; 95% ДИ: 1,1-4,0) Более высокая смертность связана с большим размером клона |
|
Abplanalp WT, et al. (2020) [24] |
Носители КГНП: 8 пациентов с тяжелым дегенеративным аортальным стенозом и 6 пациентов с СН ишемической этиологии vs 3 здоровых пациента группы-контроля Средний возраст 75,7 лет |
DNMT3A, TET2 |
Секвенирование моноцитов КГНП (TET2 + DNMT3A) vs контроль В группе КГНП наблюдалась повышенная экспрессия: ИЛ-1β, рецептора ИЛ-6, комплекса NLRP3-инфламмасома, CD163 |
|
Assmus В, et al. (2020) [49] |
419 пациентов СНнФВ преимущественно II-III ФК ишемической этиологии Медиана возраста 63 года Анализ мононуклеарных клеток костного мозга или периферической крови 56,2% случаев КГНП (DNMT3A или TET2) с VAF >0,5% Наблюдение 5 лет |
DNMT3A, TET2 |
Более высокая 5-летняя смертность среди носителей КГНП: при наличии мутации в одном из генов DNMT3A или TET2: 29% (95% ДИ: 11-46%); при наличии мутации в генах DNMT3A и TET2: 42% (95% ДИ: 26-57%) vs без КГНП: 18% (95% ДИ: 14-21%) |
|
Cremer S, et al. (2020) [52] |
419 пациентов с СНнФВ ишемической этиологии, преимущественно II ФК Медиана возраста 63 года Анализ мононуклеарных клеток костного мозга или периферической крови 36,7% случаев КГНП с VAF <2% Наблюдение 4 года |
DNMT3A, TET2, PPM1D, SRSF2 |
Более высокая смертность среди носителей КГНП Более высокая смертность, связанная с размером клона Более высокая смертность, связанная с количеством мутаций |
|
Kiefer KC, et al. (2021) [50] |
399 пациентов с СНнФВ ишемической этиологии II ФК Медиана возраста 63 года 87% случаев КГНП с VAF ≥0,5% Наблюдение 3,95 года |
DNMT3A, TET2, SRSF2 |
Мутации в CBL, CEBPA, EZH2, GNB1, PHF6, SMC1A, SRSF2 связаны с более высокой смертностью независимо от TET2/DNMT3A |
|
Palomo L, et al. (2021) [53] |
60 пациентов с СН ишемической (41,7%) и неишемической (58,3%) этиологии ФВ ЛЖ 40,1±13,4% 17 (28%) случаев с КГНП Наблюдение 3,6 года |
DNMT3A, TET2 |
Мутация DNMT3A ассоциируется с диастолической дисфункцией Отсутствие увеличения смертности среди пациентов с КГНП (ОР 1,53; 95% ДИ: 0,45-5,24) Отсутствие увеличения числа случаев СН госпитализаций и смерти среди пациентов с КГНП (ОР 2,12; 95% ДИ: 0,79-5,71) |
|
Pascual-Figal DA, et al. (2021) [51] |
62 пациента с СНнФВ Неишемическая этиология 51,6% Средний возраст 74 года 24 (38,7%) случаев с КГНП Наблюдение 3,65 года |
DNMT3A, TET2 |
Прогрессирование СН среди пациентов с DNMT3A/TET2-мутациями независимо от этиологии СН Увеличение риска смерти (ОР 2,79; 95% ДИ: 1,31-5,92) Увеличение риска смерти или госпитализации в связи с СН (ОР 3,84; 95% ДИ: 1,84-8,04) Увеличение риска смерти по причине СН или госпитализация по поводу декомпенсации СН (ОР 4,41; 95% ДИ: 2,15-9,03) |
|
Cochran J, et al. (2023) [55] |
Когорты с КГНП: 81 пациент с СНсФВ (средний возраст 71±6 лет) vs 36 добровольцев группы контроля без СНсФВ (средний возраст 74±7 лет) Наблюдение за пациентами СНсФВ 5 лет |
DNMT3A, TET2, ASXL1, TP53, SF3B1 |
Мутации TET2 достоверно чаще встречались у пациентов с СНсФВ по сравнению с группой контроля У пациентов с СНсФВ КГНП был связан с ухудшением диастолической функции сердца и увеличением числа сердечно-сосудистых госпитализаций |
Примечание: * — представлены только основные гены, обсуждаемые в статье, панель исследуемых генов может быть шире.
Сокращения: ДИ — доверительный интервал, ИЛ — интерлейкин, КГНП — клональный гемопоэз неопределенного потенциала, OP — отношение рисков, СКК — стволовые клетки костного мозга, СН — сердечная недостаточность, СНнФВ — сердечная недостаточность с низкой фракций выброса, СНсФВ — сердечная недостаточность с сохраненной фракций выброса, ФВ ЛЖ — фракция выброса левого желудочка, ФК — функциональный класс, ARIC — Atherosclerosis Risk In Communities, study, CHS — Cardiovascular Health Study, JHS — Jackson Heart Study, PREVEND — Prevention of Renal and Vascular End-stage Disease, UKBB — United Kingdom BioBank, WHI — Women's Health Initiative, VAF — Variant Allele Fraction.
Метаанализ 5 проспективных популяционных исследований с участием 56597 человек (средний возраст участников 58 лет в исходной точке наблюдения) показал, что у носителей соматических мутаций КГНП, включая мутации TET2, ASXL1, JAK2, риск развития СН в течение 20 лет повышался на 25% и не зависел от других традиционных причинных факторов. Мутация ASXL1 была ассоциирована со снижением ФВ ЛЖ, и не было получено связей СН с мутацией DNMT3A. Авторы отметили отсутствие влияния исходного наличия коронарной болезни на полученные связи КГНП с развитием СН. В то же время более высокий риск развития СН имели пациенты с большей представленностью клона (VAF >10%) [45]. В другом наблюдательном исследовании когорты Prevention of Renal and Vascular End-stage Disease (PREVEND) (медиана возраста участников 65 лет при включении) была выявлена связь КГНП с дебютом СН лишь в группе пациентов более молодого возраста (<65 лет). Индивидуальный анализ генов определил достоверные ассоциации мутации DNMT3A с развитием СН во всей исследованной когорте и фенотипом СНсФВ в группе более молодых участников. Надо отметить, что традиционные прогностические маркеры СН (натрийуретические пептиды и высокочувствительный тропонин) коррелировали с наличием КГНП в целом по группе и в группе пациентов <65 лет, в то время как в группе >65 лет корреляций с биомаркерами получено не было [46].
Одновременно накапливаются данные о связи КГНП с острыми формами СН. Продемонстрировано, что "драйверные" мутации TET2, ASXL1 чаще встречаются у пациентов с кардиогенным шоком в большинстве случаев вследствие острой декомпенсации ХСН независимо от причинного фактора по сравнению со стабильными пациентами с СН амбулаторной выборки. Заметим, что данный феномен был ассоциирован с провоспалительным цитокиновым профилем и худшей выживаемостью этих больных, как в краткосрочной перспективе, так и на протяжении трехлетнего наблюдения [47].
Вследствие того, что коронарная болезнь сердца является одной из основных причин развития ХСН, большая часть клинических работ в зоне КГНП и ХСН описана при СНнФВ ишемической этиологии. Ряд обсервационных ретроспективных исследований демонстрируют, что КГНП является возможным ФР развития СНнФВ и неблагоприятного прогноза у пациентов с постинфарктным кардиосклерозом (ПИКС) [48-50]. Опубликованные данные на сегодняшний день не дают однозначной оценки связи КГНП с возрастом пациентов СНнФВ ишемической этиологии. Рядом авторов показано, что частота выявления "драйверных" мутаций на 5-10% превышает аналогичные показатели по сравнению с обычной непрофилированной популяцией и лицами с ишемической болезнью сердца, составляя 10-27% у пациентов в возрасте 50-79 лет [7][48]. В дополнение к этому продемонстрирована высокая встречаемость мутаций КГНП с VAF ≥0,5% у пациентов с СНнФВ и ПИКС старше 50 лет независимо от возрастной категории [50].
Можем заметить, что клинические работы, посвященные прогностической значимости КГНП, в подавляющем большинстве носят ретроспективный характер. В исследовании Dorsheimer L, et al. (2019) наличие мутаций DNMT3A или TET2 у пациентов СНнФВ и ПИКС в 2 раза увеличивало риск смерти и комбинированной конечной точки (смерти или госпитализации по поводу СН) на протяжении >4 лет наблюдения, причем неблагоприятный исход был в основном обусловлен прогрессированием ХСН и аритмиями, а не острыми коронарными событиями [48]. Аналогичные данные были получены в работе Pascual-Figal DA, et al. (2021), где было продемонстрировано влияние тех же "драйверных" генов КГНП на неблагоприятное течение СНнФВ в долгосрочной перспективе независимо от её этиологии. Наличие мутаций DNMT3A или TET2 в 4 раза увеличивало риски смерти (включая внезапную сердечную смерть или смерть по причине терминальной СН) или госпитализации по причине СН даже с поправкой на такие традиционные прогностические риски, как возраст, коронарная болезнь сердца, ФВ ЛЖ, уровень натрийуретического пептида [51]. Наряду с предсказательной значимостью мутаций DNMT3A и TET2, смертность >40% на отрезке четырех лет наблюдения была показана у пациентов с СНнФВ ишемической этиологии, имеющих "драйверные" мутации SRSF2, PPM1D и других генов [52]. Эта же группа исследователей и прочие авторы сообщили о влиянии на исходы СНнФВ не только мутирующего гена, но и количества мутаций и размера мутантного клона КГНП [49][50][52]. Например, среди пациентов СН, носителей ≥2 мутаций КГНП, риск смерти увеличивался вдвое по сравнению с лицами, имеющими одну мутацию, аналогичная закономерность прослеживалась при большем размере клона [52].
В то же время анализ выборки пациентов ХСН I-III ФК и ФВ ЛЖ 40,1±13,4% (в 52% случаев больные имели неишемическую этиологию ХСН) не выявил корреляций КГНП с трехлетним прогнозом, при этом "драйверная" мутация DNMT3A ассоциировалась с диастолической дисфункцией в исходной точке исследования [53].
Нездоровое старение и широко распространенные сопутствующие заболевания вызывают хроническое системное низкоуровневое воспалительное состояние, которое способствует неблагоприятному ремоделированию миокарда и развитию СНсФВ. Воспалительные пути, лежащие в основе парадигмы большинства патофизиологических фенотипов СНсФВ, имеют поразительное сходство с воспалительными профилями, которые обусловлены КГНП [17][54]. Однако клинические данные, посвященные КГНП и СНсФВ, крайне немногочисленны. В дополнение к указанным выше исследованиям, демонстрирующим ассоциацию мутации DNMT3A с развитием СНсФВ в группе пациентов младше 65 лет и диастолической дисфункцией, следует упомянуть еще одну работу, в которой сравнивались немногочисленные когорты с КГНП и была показана достоверно более частая встречаемость мутации TET2 у пациентов с СНсФВ по сравнению с группой контроля. Более того, в исходной точке клоны КГНП, в т. ч. мутации DNMT3A/TET2, были связаны с ухудшением диастолической функции сердца и увеличением числа сердечно-сосудистых госпитализаций на протяжении пятилетнего наблюдения [55].
Продолжая обсуждение прогностической значимости КГНП, упомянем вторичный анализ исследования CANTOS (Canakinumab ANtiinflammatory Thrombosis Outcomes Study; Исследование противовоспалительных свойств канакинумаба (моноклонального антитела, нейтрализующего ИЛ-1β, являющийся, как уже обсуждалось ранее, частью воспалительного каскада NLRP3-инфламмасомы) на тромботические события). Напомним, что в течение 5 лет изучали, может ли снижение воспаления у пациентов, перенесших инфаркт миокарда и имеющих повышенный уровень высокочувствительного С-реактивного белка, снизить риски повторных ишемических событий. Последующая оценка роли маркеров КГНП на исходы в данном исследовании выявила, что у пациентов, имеющих мутацию TET2, риск основных неблагоприятных сердечно-сосудистых событий (инфаркта миокарда, ишемического инсульта или сердечно-сосудистой смерти) снижался на 62% на фоне приёма канакинумаба по сравнению с приёмом плацебо [56]. Этот факт, наряду с клонами большего размера, позволяет предположить, что определенные фенотипы пациентов с КГНП могут лучше отвечать на противовоспалительную терапию [17][56].
Растущее количество доказательств связующей роли провоспалительных цитокинов между КГНП и ССЗ обосновывает идею о том, что уменьшение воспаления путем воздействия на различные аспекты иммунологического каскада, ассоциированного с КГНП, может снизить атерогенез и риски определенных фенотипов СН и/или ее декомпенсации. Данный подход представляет интерес для клинических исследований. Рассматриваются потенциальные терапевтические мишени для супрессирования воспаления у пациентов с КГНП: ингибирование компонентов инфламмасом NLRP3 или AIM2, воспалительных путей, ассоциируемых с ними (ИЛ-1β, ИЛ-6), или использование препаратов, специфичных для мутаций КГНП [57]. Ряд клинических исследований проводится в этой зоне (NCT05177822, NCT03797001, NCT04705987), однако они не затрагивают КГНП. На текущий момент зарегистрированы 2 протокола, направленных на патологическое воспаление у пациентов с КГНП: исследование I фазы ингибитора NLRP3 селнофласта у больных с коронарным атеросклерозом и повышенными уровнями высокочувствительного С-реактивного белка, в рамках которого запланировано подысследование, сосредоточенное на людях с патогенным вариантом TET2 КГНП; и исследование II фазы по колхицину у пациентов с КГНП и ишемической СНнФВ [17].
Нельзя не коснуться обсуждения другой возраст-ассоциируемой патологии, тесно связанной с ХСН — это аритмии, и прежде всего, фибрилляции предсердий (ФП). Недавно проведенные исследования предоставляют убедительные генетические данные, подтверждающие мнение о том, что КГНП представляет собой новый ФР ФП и широкого спектра других суправентрикулярных, бради- и желудочковых аритмий [58-60]. Использовав высокочувствительное таргетное секвенирование в когорте пациентов Восточноазиатского происхождения (возраст 50-79 лет, 1004 пациента с ФП и 3341 пациент без ФП), Ahn HJ, et al. (2024) показали, что распространенность мутаций КГНП (преимущественно DNMT3A и TET2) была в 1,3-1,7 раза выше у пациентов с ФП, даже с поправкой на возраст, пол, курение, индекс массы тела, СД и артериальную гипертензию. Проведя независимый анализ 21286 субъектов с ФП биобанка Великобритании, дополнительно авторы установили, что пациенты с ФП и КГНП имеют более высокие риски неблагоприятных клинических исходов. Комбинированная конечная точка (СН, ишемический инсульт или смерть) на протяжении среднего периода наблюдения 4 года регистрировалась на 32% чаще у носителей КГНП, чем у пациентов без мутаций [59]. В другом крупном исследовании с применением полноэкзомного секвенирования 410702 участников (медиана возраста 58 лет) была показана связь КГНП с возникновением ФП (наблюдение ~11 лет), при этом наиболее значимая ассоциация прослеживалась с сплайсосомными генами и мутациями TP53 и PPM1D. Надо отметить, что геноспецифичный анализ выявил связи повышенного риска любых аритмий, особенно остановки сердца, со всеми "драйверными" генами КГНП, за исключением DNMT3A. Авторы обнаружили ассоциацию КГНП с большим объёмом фиброза по данным магнитно-резонансного исследования (Т1-картирование) миокарда; эта связь была сильнее при увеличении размера клона и наличии "драйверного" гена TET2 [60].
Как уже упоминалось, КГНП тесно ассоциирован с другим состоянием — раком, что позволяет позиционировать этот феномен как связующее звено между онкологическими и ССЗ [61]. Многочисленные эпидемиологические данные свидетельствуют о повышенном риске развития рака у пациентов с СН и наоборот, — СН у онкологических больных, подтверждая взаимовлияние и общность ряда моментов патогенеза при этих патологиях. Для обоих состояний характерны одинаковые ФР, включая старение, нездоровый образ жизни, ожирение и СД, а также основные патофизиологические механизмы: нейрогормональная активация, метаболическое ремоделирование, хроническое воспаление и КГНП [62-64]. В связи с вышеуказанным, а также учитывая высокую распространенность КГНП у онкологических пациентов, ранее проходивших лечение, признается целесообразность дальнейших мультидисциплинарных исследований для лучшего понимания общности патогенеза КГНП, рака и ССЗ, что существенно может расширить сферу кардиоонкологии в аспекте персонализированного управления сердечно-сосудистыми рисками [61][65].
И наконец, хотелось бы отметить, что современные пациенты с ХСН российской популяции имеют средний возраст 65 лет и высокое бремя сопутствующих заболеваний, включая нозологии, ассоциируемые с КГНП [66]. Стоит подчеркнуть, что молекулярно-генетические маркеры вносят незначительный вклад в прогноз заболеваний полигенной этиологии, а этнические особенности здорового образа жизни, экологические и социальные риски потенциально способны оказывать влияние на представленность молекулярных маркеров КГНП. Вышеизложенное подчеркивает актуальность проведения национальных генетических исследований в этой области, в т. ч. на российской выборке пациентов.
Оценивая текущие данные, можно констатировать, что понимание механизмов КГНП значительно расширилось за последние 5 лет, но все же эта область научных интересов продолжает пополняться новыми фактами. КГНП представляет собой распространенное и, возможно, неизбежное последствие старения. Носители мутировавших клонов подвергаются повышенному риску не только онкологических заболеваний, но и неблагоприятным сердечно-сосудистым событиям. Современные исследования дают убедительные основания полагать, что КГНП является биомаркером развития и прогрессирования ХСН независимо от её этиологии, однако клиническая реализация данного феномена у мультиморбидных пациентов в долгосрочной перспективе на фоне современной болезнь-модифицируемой терапии требует уточнения.
В настоящее время для людей с КГНП не существует методов лечения, которые могли бы снизить риск онкопатологии или ССЗ. Это определяет вектор направления фундаментальных и трансляционных исследований для выявления генно-специфических механизмов заболеваний и определения таргетных подгрупп пациентов. В этой связи не вызывает сомнения, что КГНП и ассоциируемые с ним воспалительные пути представляют собой мощную терапевтическую мишень, что обусловливает актуальность научных работ в зоне различных стадий ХСН и маркеров КГНП, потенциально подходящих для иммунотерапевтических препаратов.
В целом, можно заключить, что понимание сложных взаимодействий между мутантными клонами, иммунными путями и хроническим воспалением будет иметь важное значение в разработке алгоритмов персонифицированного подхода профилактических, терапевтических и реабилитационных мероприятий у людей с КГНП.
Отношения и деятельность. Исследование выполнено при финансовой поддержке Министерства науки и высшего образования Российской Федерации (соглашение № 075-15-2022-301 от 20.04.2022).
1. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/ American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895-e1032. doi:10.1161/CIR.0000000000001063.
2. Murphy SP, Kakkar R, McCarthy CP, et al. Inflammation in Heart Failure: JACC State-ofthe-Art Review. J Am Coll Cardiol. 2020;75(11):1324-40. doi:10.1016/j.jacc.2020.01.014.
3. Rauchhaus M, Doehner W, Francis DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102(25):3060-7. doi:10.1161/01.cir.102.25.3060.
4. Florez MA, Tran BT, Wathan TK, et al. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell. 2022;29(6):882-904. doi:10.1016/j.stem.2022.05.006.
5. Haring B, Wissel S, Manson JE. Somatic Mutations and Clonal Hematopoiesis as Drivers of Age-Related Cardiovascular Risk. Curr Cardiol Rep. 2022;24(8):1049-58. doi:10.1007/s11886-022-01724-2.
6. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. doi:10.1182/blood-2015-03-631747.
7. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-98. doi:10.1056/NEJMoa1408617.
8. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111-21. doi:10.1056/NEJMoa1701719.
9. Mas-Peiro S, Hoffmann J, Fichtlscherer S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. 2020;41(8):933-9. doi:10.1093/eurheartj/ehz591.
10. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018; 10(436):eaan8292. doi:10.1126/scitranslmed.aan8292.
11. Kimishima Y, Misaka T, Yokokawa T, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun. 2021;12(1):6177. doi:10.1038/s41467-021-26435-0.
12. Jensen JL, Easaw S, Anderson T, et al. Clonal Hematopoiesis and the Heart: a Toxic Relationship. Curr Oncol Rep. 2023;25(5):455-63. doi:10.1007/s11912-023-01398-1.
13. Слепцов А.А., Назаренко М. С., Пузырев В. П. Общее в атеро и канцерогенезе: клональный гемопоэз. Российский кардиологический журнал. 2023;28(10):5511. doi:10.15829/1560-4071-2023-5511.
14. Bick AG, Pirruccello JP, Griffin GK, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 2020;141(2):124-31. doi:10.1161/CIRCULATIONAHA.119.044362.
15. Mayerhofer E, Strecker C, Becker H, et al. Prevalence and Therapeutic Implications of Clonal Hematopoiesis of Indeterminate Potential in Young Patients With Stroke. Stroke. 2023;54(4):938-46. doi:10.1161/STROKEAHA.122.041416.
16. Bick AG, Weinstock JS, Nandakumar SK, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586(7831):763-8. doi:10.1038/s41586-020-2819-2.
17. Sikking MA, Stroeks SLVM, Waring OJ, et al. Clonal Hematopoiesis of Indeterminate Potential From a Heart Failure Specialist's Point of View. J Am Heart Assoc. 2023;12 (15):e030603. doi:10.1161/JAHA.123.030603.
18. Stacey SN, Zink F, Halldorsson GH, et al. Genetics and epidemiology of mutational barcode-defined clonal hematopoiesis. Nat Genet. 2023;55(12):2149-59. doi:10.1038/s41588-023-01555-z.
19. Kandarakov O, Belyavsky A. Clonal Hematopoiesis, Cardiovascular Diseases and Hematopoietic Stem Cells. Int J Mol Sci. 2020;21(21):7902. doi:10.3390/ijms21217902.
20. Challen GA, Goodell MA. Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood. 2020;136(14):1590-8. doi:10.1182/blood.2020006510.
21. Avagyan S, Henninger JE, Mannherz WP, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science. 2021;374(6568):768-72. doi:10.1126/science.aba9304.
22. Zhang Q, Zhao K, Shen Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389-93. doi:10.1038/nature15252.
23. Sano S, Oshima K, Wang Y, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J Am Coll Cardiol. 2018;71(8):875-86. doi:10.1016/j.jacc.2017.12.037.
24. Abplanalp WT, Cremer S, John D, et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res. 2021;128(2):216-28. doi:10.1161/ CIRCRESAHA.120.317104.
25. Sano S, Oshima K, Wang Y, et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 2018;123(3):335-41. doi:10.1161/CIRCRESAHA.118.313225.
26. Sano S, Wang Y, Yura Y, et al. JAK2V617F-Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci. 2019;4(6): 684-97. doi:10.1016/j.jacbts.2019.05.013.
27. Fidler TP, Xue C, Yalcinkaya M, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296-301. doi:10.1038/s41586-021-03341-5.
28. Jaiswal S, Libby P.Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17(3):137-44. doi:10.1038/s41569-019-0247-5.
29. Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood. 2020;136(14): 1606-14. doi:10.1182/blood.2019000989.
30. Wang W, Liu W, Fidler T, et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 V617F Mice. Circulation Research. 2018;123:e35- e47. doi:10.1161/circresaha.118.313283.
31. Abplanalp WT, Mas-Peiro S, Cremer S, et al. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. 2020;5:1170-5. doi:10.1001/jamacardio.2020.2468.
32. Sano S, Wang Y, Ogawa H, et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophilmediated cytotoxic response. JCI Insight. 2021 Jul 8;6(13):e146076. doi:10.1172/jci.insight.146076.
33. Yura Y, Miura-Yura E, Katanasaka Y, et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ Res. 2021;129(6):684-98. doi:10.1161/CIRCRESAHA.121.319314.
34. Liao M, Chen R, Yang Y, et al. Aging-elevated inflammation promotes DNMT3A R878Hdriven clonal hematopoiesis. Acta Pharm Sin B. 2022;12:678-91. doi:10.1016/j.apsb.2021.09.015.
35. Abegunde SO, Buckstein R, Wells RA, et al. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp Hematol. 2018;59:60-5. doi: 10.1016/j.exphem.2017.11.002.
36. Yura Y, Sano S, Walsh K. Clonal Hematopoiesis: A New Step Linking Inflammation to Heart Failure. JACC Basic Transl Sci. 2020;5(2):196-207. doi:10.1016/j.jacbts.2019.08.006.
37. Abplanalp WT, Schuhmacher B, Cremer S, et al. Cell-intrinsic effects of clonal hematopoiesis in heart failure. Nat Cardiovasc Res. 2023;2:819-34. doi:10.1038/s44161-023-00322-x.
38. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366(6465):eaan4673. doi:10.1126/science.aan4673.
39. Piepoli MF, Adamo M, Barison A, et al. Preventing heart failure: a position paper of the Heart Failure Association in collaboration with the European Association of Preventive Cardiology. Eur J Prev Cardiol. 2022;29(1):275-300. doi:10.1093/eurjpc/zwab147.
40. Van Deuren RC, Andersson-Assarsson JC, Kristensson FM, et al. Expansion of mutationdriven haematopoietic clones is associated with insulin resistance and low HDL-cholesterol in individuals with obesity. bioRxiv, 2021.05.12.443095. doi:10.1101/2021.05.12.443095.
41. Miller PG, Qiao D, Rojas-Quintero J, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. 2022;139(3):357-68. doi:10.1182/blood.2021013531.
42. Vlasschaert C, McNaughton AJM, Chong M, et al. Association of Clonal Hematopoiesis of Indeterminate Potential with Worse Kidney Function and Anemia in Two Cohorts of Patients with Advanced Chronic Kidney Disease. J Am Soc Nephrol. 2022;33(5):985-95. doi:10.1681/ASN.2021060774.
43. Komic L, Kumric M, Urlic H, et al. Obesity and Clonal Hematopoiesis of Indeterminate Potential: Allies in Cardiovascular Diseases and Malignancies. Life (Basel). 2023;13(6):1365. doi:10.3390/life13061365.
44. Hatakeyama K, Hieda M, Semba Y, et al. TET2 Clonal Hematopoiesis Is Associated With Anthracycline-Induced Cardiotoxicity in Patients With Lymphoma. JACC CardioOncol. 2022;4(1):141-3. doi:10.1016/j.jaccao.2022.01.098.
45. Yu B, Roberts MB, Raffield LM, et al. National Heart, Lung, and Blood Institute TOP Med Consortium. Supplemental Association of Clonal Hematopoiesis with Incident Heart Failure. J Am Coll Cardiol. 2021;78(1):42-52. doi:10.1016/j.jacc.2021.04.085.
46. Shi C, Aboumsallem JP, Suthahar N, et al. Clonal hematopoiesis of indeterminate potential: associations with heart failure incidence, clinical parameters and biomarkers. Eur J Heart Fail. 2023;25(1):4-13. doi:10.1002/ejhf.2715.
47. Scolari FL, Abelson S, Brahmbhatt DH, et al. Clonal haematopoiesis is associated with higher mortality in patients with cardiogenic shock. Eur J Heart Fail. 2022;24(9):1573-82. doi:10.1002/ejhf.2588.
48. Dorsheimer L, Assmus B, Rasper T, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019; 4(1):25-33. doi:10.1001/jamacardio.2018.3965.
49. Assmus B, Cremer S, Kirschbaum K, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur Heart J. 2021;42(3):257-65. doi:10.1093/eurheartj/ehaa845.
50. Kiefer KC, Cremer S, Pardali E, et al. Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality. ESC Heart Fail. 2021;8(3):1873-84. doi:10.1002/ehf2.13297.
51. Pascual-Figal DA, Bayes-Genis A, Díez-Díez M, et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. J Am Coll Cardiol. 2021;77(14):1747-59. doi:10.1016/j.jacc.2021.02.028.
52. Cremer S, Kirschbaum K, Berkowitsch A, et al. Multiple Somatic Mutations for Clonal Hematopoiesis Are Associated With Increased Mortality in Patients With Chronic Heart Failure. Circ Genom Precis Med. 2020;13(4):e003003. doi:10.1161/CIRCGEN.120.003003.
53. Palomo L, Santiago-Vacas E, Pascual-Figal D, et al. Prevalence and characteristics of clonal hematopoiesis in heart failure. Rev Esp Cardiol (Engl Ed). 2021;74(11):996-9. English, Spanish. doi:10.1016/j.rec.2021.05.005.
54. Pugliese NR, Pellicori P, Filidei F, et al. Inflammatory pathways in heart failure with preserved left ventricular ejection fraction: implications for future interventions. Cardiovasc Res. 2023;118(18):3536-55. doi:10.1093/cvr/cvac133.
55. Cochran JD, Yura Y, Thel MC, et al. Clonal Hematopoiesis in Clinical and Experimental Heart Failure With Preserved Ejection Fraction. Circulation. 2023;148(15):1165-78. doi:10.1161/CIRCULATIONAHA.123.064170.
56. Svensson EC, Madar A, Campbell CD, et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022;7(5):521-8. doi:10.1001/jamacardio.2022.0386.
57. Tall AR, Fuster JJ. Clonal hematopoiesis in cardiovascular disease and therapeutic implications. Nat Cardiovasc Res. 2022;1(2):116-24. doi:10.1038/s44161-021-00015-3.
58. Zuriaga MA, Pascual-Figal D, Fuster JJ. Clonal haematopoiesis and cardiac arrythmias: rhythm-altering mutations. Eur Heart J. 2024;45(10):806-8. doi:10.1093/eurheartj/ehae052.
59. Ahn HJ, An HY, Ryu G, et al. Clonal haematopoiesis of indeterminate potential and atrial fibrillation: an east Asian cohort study. Eur Heart J. 2024;45(10):778-90. doi:10.1093/eurheartj/ehad869.
60. Schuermans A, Vlasschaert C, Nauffal V, et al. Clonal haematopoiesis of indeterminate potential predicts incident cardiac arrhythmias. Eur Heart J. 2024;45(10):791-805. doi:10.1093/eurheartj/ehad670.
61. Libby P, Sidlow R, Lin AE, et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74(4):567-77. doi:10.1016/j.jacc.2019.06.007.
62. de Boer RA, Hulot JS, Tocchetti CG, et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2020;22(12):2272-89. doi:10.1002/ejhf.2029.
63. Aboumsallem JP, Moslehi J, de Boer RA. Reverse Cardio-Oncology: Cancer Development in Patients With Cardiovascular Disease. J Am Heart Assoc. 2020;9(2):e013754. doi:10.1161/JAHA.119.013754.
64. Kadowaki H, Akazawa H, Shindo A, et al. Shared and Reciprocal Mechanisms Between Heart Failure and Cancer - An Emerging Concept of Heart-Cancer Axis. Circ J. 2024; 88(2):182-8. doi:10.1253/circj.CJ-23-0838.
65. Stein A, Metzeler K, Kubasch AS, et al. Clonal hematopoiesis and cardiovascular disease: deciphering interconnections. Basic Res Cardiol. 2022;117(1):55. doi:10.1007/s00395-022-00969-w.
66. Шляхто Е.В., Беленков Ю.Н., Бойцов С.А. и др. Результаты промежуточного анализа проспективного наблюдательного многоцентрового регистрового исследования пациентов с хронической сердечной недостаточностью в Российской Федерации "ПРИОРИТЕТХСН": исходные характеристики и лечение первых включенных пациентов. Российский кардиологический журнал. 2023;28(10):5593. doi:10.15829/1560-4071-2023-5593.
Лясникова Е. А. - к.м.н., доцент кафедры факультетской терапии с клиникой Института медицинского образования, зав. НИЛ предиабета и метаболических нарушений НЦМУ "Центр персонализированной медицины"
Санкт-Петербург
AuthorID: 630408
Иванченко Л. Ю. - лаборант-исследователь НИЛ предиабета и метаболических нарушений НЦМУ "Центр персонализированной медицины", аспирант кафедры факультетской терапии с клиникой Института медицинского образования
Санкт-Петербург
AuthorID: 1222614
Козлова С. Н. - д.м.н., профессор кафедры факультетской терапии с клиникой Института медицинского образования
Санкт-Петербург
AuthorID: 657957
Ситникова М. Ю. - д.м.н., г.н.с., зав. НИО сердечной недостаточности, профессор кафедры факультетской терапии c клиникой Института медицинского образования
Санкт-Петербург
AuthorID: 630219
Костарева А.А. - д.м.н., директор Института молекулярной биологии и генетики, профессор кафедры факультетской терапии с клиникой Института медицинского образования
Санкт-Петербург
AuthorID: 610181
Шляхто Е.В. - д.м.н., академик РАН, генеральный директор, зав. кафедрой факультетской терапии с клиникой Института медицинского образования
Санкт-Петербург
AuthorID: 136786
Лясникова Е.А., Иванченко Л.Ю., Козлова С.Н., Ситникова М.Ю., Костарева А.А., Шляхто Е.В. Клональный гемопоэз неопределённого потенциала и хроническая сердечная недостаточность. Российский кардиологический журнал. 2024;29(11S):6016. https://doi.org/10.15829/1560-4071-2024-6016
Lyasnikova E.A., Ivanchenko L.Yu., Kozlova S.N., SITNIKOVA M.Yu., Kostareva A.A., Shlyakhto E.V. Clonal hematopoiesis of indeterminate potential and heart failure. Russian Journal of Cardiology. 2024;29(11S):6016. (In Russ.) https://doi.org/10.15829/1560-4071-2024-6016













































