



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2024-5986
Перейти к:
В патогенезе многих воспалительных процессов важную роль играет каскад реакций различных видов инфламмасом. Продуктами их активации выступают провоспалительные цитокины интерлейкин (IL)-1β и IL-18. Эти белковые молекулы могут секретироваться двумя различными способами: везикулярным транспортом либо посредством образования пор в клеточной мембране, что в дальнейшем ведет к гибели секретирующей клетки. Роль активации инфламмасом в клетках сердечной ткани на настоящее время изучена недостаточно, однако есть некоторые исследования, отражающие связь запуска инфламмасомного каскада с развитием сердечно-сосудистых заболеваний. Таким образом, активация инфламмасом в кардиомиоцитах может приводить к электролитному дисбалансу, что впоследствии ведет к образованию эктопических очагов в сердечной ткани и нарушению сердечного ритма. Запуск инфламмасомного каскада в сердечных фибробластах способствует формированию фиброза и ремоделированию ткани миокарда, что приводит к нарушению функциональной активности сердца. Активация инфламмасомы в эндотелиоцитах коронарных артерий приводит к эндотелиальной дисфункции и атерогенезу. Таким образом, активация различных видов инфламмасом в сердечной ткани приводит к формированию сердечной патологии.
Рубинштейн А.А., Ходот А.А., Тирикова П.В., Головкин А.С., Кудрявцев И.В., Шляхто Е.В. Инфламмасома - новый взгляд на терапию сердечно-сосудистых заболеваний: обзор. Часть I. Российский кардиологический журнал. 2024;29(11S):5986. https://doi.org/10.15829/1560-4071-2024-5986
Rubinstein A.A., Khodot A.A., Tirikova P.V., Golovkin A.S., Kudryavtsev I.V., Shlyakhto E.V. Inflammasome - a new look at the therapy of cardiovascular diseases: a review. Part I. Russian Journal of Cardiology. 2024;29(11S):5986. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5986
Воспаление как типовой патологический процесс является обязательным звеном патогенеза многих заболеваний, в т. ч. сердечно-сосудистых. Неконтролируемое местное воспаление, а также системный воспалительный ответ оказываются триггером каскадов реакций, приводящих к более тяжелому течению основного заболевания, развитию осложнений, неблагоприятным исходам. Важными регуляторами процессов, связанных с развитием воспаления, являются цитокины. Продукция провоспалительных цитокинов может быть инициирована как присутствием образов чужеродности патогенов, так и появлением паттернов повреждения, связанных с молекулами собственных разрушенных клеток. В результате идентификации образов патоген-распознающими рецепторами клеток запускается каскад внутриклеточных реакций, одним из результатов которого оказывается сборка инфламмасомы, осуществляющей продукцию провоспалительных цитокинов. Целью нашего обзора являлось выявление роли различного вида инфламмасом в сердечной ткани.
Методологический подход включал в себя систематический анализ оригинальных исследований, метаанализов, а также обзорных статей, опубликованных в международных базах данных ("Medline", "PubMed", "Scopus"). Ключевые слова, которые мы использовали: "инфламмасома", "NLRP3", "AIM2", "NLRC4", "кардиомиоциты", "сердечно-сосудистые заболевания". Описательный обзор проводился в соответствии с протоколом PRISMA (http://www.prisma-statement.org), используемым для этого типа исследования (ID-423604).
Инфламмасома представляет собой мультибелковый комплекс, локализованный в цитозоле клетки, при активации которого запускается каскад воспалительных реакций. Этот макромолекулярный комплекс обычно включает сенсорный белок, воспалительные каспазы и в некоторых случаях связывающий их адаптерный белок [1]. Основными продуктами инфламмасомы выступают секретируемые в межклеточное пространство провоспалительные цитокины интерлейкин (IL)-1β и IL‑18, которые синтезируются в клетке в виде белков-предшественников, которые накапливаются в цитозоле и расщепляются до активных форм под действием каспазы в результате сборки инфламмасомы [2].
В качестве сенсора, в зависимости от различных видов инфламмасом, могут выступать паттерн-распознающие белки, относящиеся к семейству NOD-подобных рецепторов (NLR, от англ. Nod-like receptor), такие как: NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRC4 и NAIP, а также молекулы, принадлежащие к семейству PYHIN белков (от англ. Pyrin and HIN domain proteins) — белковые молекулы, содержащие домены пирина (PYD) и HIN200 (от англ. hematopoietic interferon-inducible nuclear antigens with 200 amino acid repeats), к которым относятся AIM2 и IFI16 [3] (рис. 1 А). Все члены семейства NLR содержат нуклеотидсвязывающий домен (NBD, от англ. nucleotide-binding domain), С-концевой богатый лейцином повтор (LRR, от англ. leucine-rich repeat) и могут содержать либо PYD, либо домен активации и рекрутирования каспазы (CARD, от англ. caspase activating and recruitment domain), хотя в некоторых инфламмасомах они могут встречаться вместе [3]. Из этого семейства выделяется белок NAIP, который помимо NBD и LRR содержит домен, подобный бакуловирусному ингибирующему повтору (BIR) [4]. Члены семейства PYHIN характеризуются еще и наличием домена HIN200, который участвует в связывании лигандов [3]. Вышеперечисленные сенсорные белки являются паттерн-распознающими рецепторами системы врожденного иммунитета (PRR, от англ. pattern recognition receptor) и экспрессируются во многих видах клеток, включая макрофаги, дендритные клетки, нейтрофилы, эпителиальные клетки различного происхождения [5]. Результаты последних лет показывают, что помимо клеток миелоидного ряда, PRR экспрессируются клетками различных тканей [6], в т. ч. кардиомиоцитами, фибробластами и эндотелиальными клетками сердечной ткани [7-9].
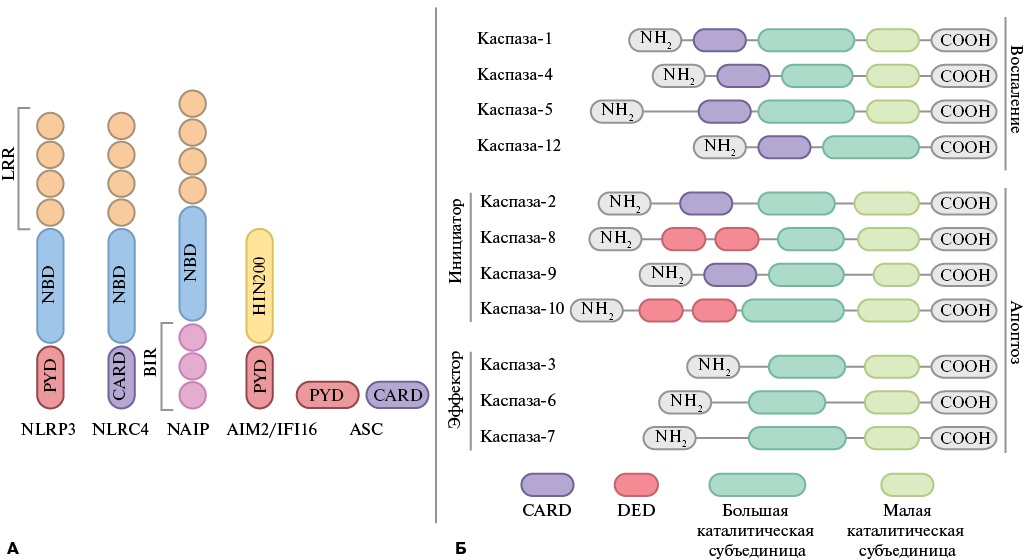
Рис. 1. А — строение белков-сенсоров: С-концевой богатый лейцином повтор (LRR); нуклеотидсвязывающий домен (NBD); белковые молекулы, содержащие домены пирина (PYD); домен активации и рекрутирования каспазы (CARD); домен, подобный бакуловирусному ингибирующему повтору (BIR); Б — различные виды каспаз. Воспалительные каспазы участвуют в запуске провоспалительного ответа как посредством процессинга про‑IL‑1β и про‑IL‑18, так и при помощи регуляции транскрипции генов цитокинов. Апоптопические каспазы подразделяются на каспазы-инициаторы, расщепляющие предшественников каспаз-эффекторов и каспазы-эффекторы, непосредственно запускающие апоптоз.
Адаптерным белком выступает ассоциированный с апоптозом пятнышкообразный белок, содержащий CARD (ASC) [10], однако он входит в состав не всех видов инфламмасом. Например, у NLRP3/6/7, AIM2, IFI16‑инфламмасом он участвует в сборке инфламмасомы, тогда как для сборки NLRC4‑инфламмасомы его участие не требуется [11] (рис. 1 А).
Воспалительные каспазы в клетках человека представлены следующими типами — каспаза‑1, каспаза‑4, каспаза‑5 и каспаза‑12 [12]. Запуск инфламмасомы, активирующий каспазу-1, считается каноническим при воспалении, а активация каспазы-4/5/12 — неканонической [1]. Эти ферменты синтезируются в клетке в виде форм-предшественников — прокаспаз, для активации которых необходимо взаимодействие между CARD‑доменом самой прокаспазы и CARD‑доменом белка-адаптера (ASC) или же CARD‑доменом белка-сенсора в тех инфламмасомах, запуск каскада реакций которых происходит за счет ASC‑независимого механизма [13]. За счет этого взаимодействия происходит саморасщепление и активация каспазы. Существуют также каспазы, вызывающие апоптоз клетки. Их условно можно подразделить на каспазы-инициаторы, которые необходимы для расщепления предшественников каспаз-эффекторов, и каспазы-эффекторы, главная функция которых заключается непосредственно в инициации апоптоза (рис. 1 Б) [14].
Запуск сборки инфламмасомы осуществляется в два этапа — "праймирование" и активация [2]. Праймирование начинается с распознавания PRR различных PAMP (от англ. pathogen-associated molecular pattern), включающих эндо- и экзотоксины бактерий, грибов, паразитов и вирусов, и DAMP (от англ. damage-associated molecular pattern), представляющих собой сигналы опасности хозяина, которые высвобождаются при повреждении клеток и тканей. Поскольку конститутивный уровень экспрессии сенсорных белков, например, NLRP3, недостаточен для активации и сборки инфламмасомы [15], клетка должна сначала распознать PAMP или DAMP при помощи TLRs, NLRs или других PRRs, что приведёт к усилению трансляционной регуляции белка-сенсора (NLRP3 и других), прокаспазы-1 [16] и про‑IL‑1β, про‑IL‑18 через активацию пути фактора транскрипции NF‑κB [17]. Таким образом, на этапе "праймирования" происходит увеличение экспрессии ключевых компонентов инфламмасомы. Тогда как на этапе активации клетки PAMPs и/или DAMPs, взаимодействуя непосредственно с сенсорными белками (NLRP3, AIM2, NLRC4 и т. п.), стимулируют сборку активного комплекса инфламмасомы с последующим запуском каскада её реакций. На данном этапе в качестве индукторов запуска инфламмасомы могут выступать также кристаллы мочевой кислоты, изменения электролитного состава клетки (снижение цитозольных концентраций ионов К+, увеличение притока ионов Na+ в клетку) [2], активные формы кислорода [18]. Также было показано, что после оттока K+ из клетки активируется связанная с митозом серин-треониновая киназа (NEK7), вызывающая олигомеризацию и активацию NLRP3‑инфламмасомы [19] (табл. 1).
Таблица 1
Паттерны, распознающиеся белками-сенсорами различных видов инфламмасом
|
Белок-сенсор |
PAMP |
DAMP |
|
NLRP3 |
Липополисахарид, компоненты пептидогликана, порообразующие токсины, вирусные нуклеиновые кислоты (например, у вирусов семейства Picornaviridae, включающие Cardiovirus A, семейства Adenoviridae, Orthomyxoviridae, Retroviridae, Hepadnaviridae), грибковые патогены (в частности компоненты клеточных стенок Candida albicans и Aspergillus fumigatus), паразитарные патогены (Schistosoma mansoni и Dermatophagoides pteronyssinus) |
Выход ионов К+, Cl-, увеличение концентрации Na+, Ca2+ в цитозоле, активация P2X7, АТФ, кристаллы гидроксида алюминия, кристаллы мочевой кислоты, кристаллы холестерина, митохондриальные активные формы кислорода, митохондриальная ДНК, пептиды повреждения лизосом, кардиолипин, гиалуронан |
|
AIM2 |
Вирусные ДНК (например, цитомегаловирус, вирус Эпштейна-Барр, вирус коровьей оспы, папилломавирус), ДНК внутриклеточных бактерий (например, Francisella tularensis, Listeria monocytogenes, Streptococcus Pneumoniae, Legionella pneumophila и Staphylococcus aureus) |
Цитозольная ДНК клеток (измененные или неправильно локализованные молекулы ДНК: поврежденная ДНК или геномная ДНК, высвобождаемая в цитозоль при потере целостности ядерной оболочки, секретируемая экзосомами ДНК соседних клеток) |
|
NLRC4 |
Флагеллин, белки систем бактериальной секреции III и IV типов, другие компоненты клеточных стенок факультативных внутриклеточных бактерий (например, Salmonella Typhimurium, Escherichia coli, Shigella flexneri, Pseudomonas aeruginosa, Burkholderia thailandensis, Chromobacterium violaceum и Legionella pneumophila) |
Cтресс-опосредованная активация p53 |
Активированные сенсорные белки, представляющие собой паттерн-распознающие рецепторы, объединяются в макромолекулярные комплексы в форме "колеса", которые инициируют олигомеризацию филаментов, образованных белком-адаптером ASC. CARD-субъединица ASC привлекает в состав комплекса прокаспазу-1, взаимодействуя с CARD-субъединицей последней, чем вызывает аутоактивацию каспазы [13]. Как было отмечено ранее, для некоторых видов инфламмасом белок-адаптер не требуется. Например, NLRC4‑инфламмасома рекрутирует прокаспазу-1 за счет взаимодействия CARD-субъединицы непосредственно белка NLRC4 с CARD-субъединицей прокаспазы [1]. Активная каспаза‑1 расщепляет цитокины проформы IL‑1β и IL‑18 до их биологически активных форм [20]. Кроме того, активированная каспаза‑1 также расщепляет газдермин D (GSDMD, от англ. Gasdermin D), что позволяет N-концевому домену GSDMD образовывать поры в цитоплазматической мембране клетки. При этом GSDMD-индуцированная пора обеспечивает высвобождение IL‑1β и IL‑18, а также ведёт к "набуханию" клетки, вызывая её провоспалительную форму гибели, называемую "пироптозом" [21]. Однако стоит отметить, что секреция IL‑1β и IL‑18 в клетках миелоидного происхождения осуществляется также через везикулярный транспорт, что способствует долгосрочному выделению этих цитокинов без индукции пироптоза в клетке-продуценте [22][23].
Цитокины IL‑1β и IL‑18 играют важную роль в поддержании воспаления как на местном, так и на системном уровне. Например, IL‑18 стимулирует продукцию IFNγ, усиливает цитолитические свойства Т- и NK-клеток, усиливает миграцию нейтрофилов в очаг воспаления, способствует секреции других провоспалительных цитокинов, включая TNFα, IL‑1β, IL‑8 и GM-CSF [24][25]. В свою очередь, IL‑1β способствует повышению температуры тела, расширению сосудов, а также привлечению различных иммунных клеток в очаг воспаления (например, Th17) [24]. Таким образом, активация инфламмасомы в клетках различного происхождения способствует секреции провоспалительных цитокинов IL‑1β и IL‑18, что может поддерживать воспалительные реакции организма и вызывать пироптоз некоторых клеток.
Помимо клеток врожденного иммунитета кардиомиоциты, фибробласты сердечной ткани, а также эндотелиальные клетки коронарных артерий способны к экспрессии различных типов инфламмасом [26-28]. Как было сказано ранее, компоненты инфламмасомы, за исключением цитозольных паттерн-распознающих рецепторов, в этих клетках не экспрессируются на постоянной основе, а являются индуцибельными, т. к. их синтез регулируется запуском фактора транскрипции NF‑κB [1][29][30]. При различных патологических процессах в сердце, включая ишемию, воспаление, нарушение ритма, ремоделирование миокарда, поражение коронарных артерий, происходит активация инфламмасом в клетках сердечной ткани. Было показано, что для синтеза компонентов инфламмасомы с их последующей активацией необходимо либо взаимодействие PAMPs и/или DAMPs с паттерн-распознающими рецепторами, либо нарушение электролитного баланса в кардиомиоцитах, которое проявляется в виде изменения цитозольных концентраций ионов калия (K+), кальция (Ca2+), хлора (Cl-), натрия (Na+) [1][2]. Для активации фактора транскрипции NF‑κB необходимо взаимодействие вышеперечисленных факторов с Toll-подобными рецепторами [1]. Известно, что в сердечной ткани человека экспрессируется широкий спектр распознающих рецепторов системы врожденного иммунитета, включая TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9 и TLR10 [31][32], лигандами для которых могут выступать PAMPs различных инфекционных агентов, а также DAMPs собственных поврежденных клеток. Таким образом, при повреждении кардиомиоцитов, нарушении их электролитного состава, а также при инфекционном поражении клеток ткани сердца происходит активация Toll-подобных рецепторов с последующим запуском NF‑κB, стимулирующим сборку инфламмасомы, что, по-видимому, является одной из ключевых причин развития воспалительного процесса.
В настоящее время известно, что в клетках сердечной ткани человека могут активироваться различные типы инфламмасом: NLRP3 [33], NLRP12 [34], NLRC4 [35], AIM2 [36], NOD2 и NLRC3 [37] (рис. 2). Запуск инфламмасомы в кардиомиоцитах может вызвать их гибель пироптозом за счет образования поры газдермином D, которая необходима для секреции IL‑1β и IL‑18 во внеклеточное пространство [7][38]. Более того, при активации NLRP3 инфламмасомы в сердечных фибробластах инициируются комплексы реакций, сопровождающиеся развитием фиброза [8]. В основе этого процесса может находиться активация каспазы-1, которая приводит к усилению секреции коллагена фибробластами, а также их дифференцировка в сторону миофибробластов [39][40]. Также активно обсуждается теория об аутокринной регуляции с формированием петли обратной усилительной связи, в основе которой находится взаимодействие рецепторов к IL‑1 на поверхности фибробластов с секретируемым IL‑1β, являющимся основным продуктом активации инфламмасомы [41]. Данное взаимодействие помимо поддержания воспаления способствует усилению синтеза и отложению коллагена в межклеточном пространстве. Стоит также отметить, что фибробласты способны к долгосрочной продукции IL‑1β в ответ на провоспалительные стимулы [8]. При этом механизм секреции фибробластами и миофибробластами цитокинов, являющихся продуктами активации инфламмасом, не связан с образованием пор газдермином D. Можно предполагать, что секреция IL‑1β и IL‑18 идет по механизмам, описанным для тканевых макрофагов, и осуществляется при помощи везикулярного транспорта [22][23]. Также есть сообщения об альтернативном механизме развития фиброза, посредством которого NLRP3 модулирует выход активных форм кислорода из митохондрий, усиливая передачу сигналов Smad и, как следствие, увеличивая экспрессию "профибротических" генов в сердечных фибробластах [8]. Таким образом, запуск инфламмасомы в кардиомиоцитах и сердечных фибробластах способствует фиброзированию миокарда и сопровождается ремоделированием ткани сердца.
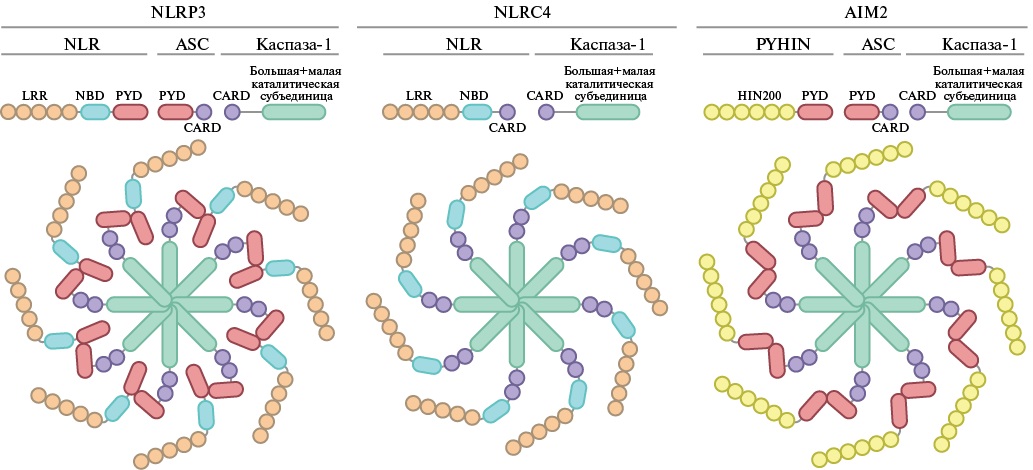
Рис. 2. Строение различных видов инфламмасом.
При гибели кардиомиоцитов пироптозом или же посредством любых других механизмов, запускаемых ишемией или инфекционными агентами, будут высвобождаться многочисленные DAMPs в межклеточное пространство. Как уже отмечалось ранее, эти DAMPs способны запускать каскад инфламмасом как в близлежащих кардиомиоцитах, вызывая их гибель, так и в сердечных фибробластах, способствуя фиброзу и ремоделированию сердечной ткани. Активация инфламмасомы в кардиомиоцитах может вызывать эктопическую активность. Так, при фибрилляции предсердий кардиомиоциты экспрессировали активированную NLRP3‑инфламмасому, которая способствовала повышенному высвобождению Ca2+ из эндоплазматического ретикулума и укорочению рефрактерного периода кардиомиоцитов предсердий [42]. Это может быть обусловлено аутокринным механизмом, заключающимся во взаимодействии IL‑1β со специфическими рецепторами на мембране кардиомиоцитов. Например, при стимуляции кардиомиоцитов данным цитокином повышается экспрессия Ca2+/кальмодулин-зависимой протеинкиназы-II (CaMKII), канала рианодинового рецептора типа 2 (RyR2), а также катализируется CaMKII-зависимое фосфорилирование сердечного RyR2 [43]. Это может вызывать триггерную активность, индуцирующую эктопические очаги. Механизм ее основан на том, что выход Ca2+ из эндоплазматического ретикулума вызывает раннюю и/или отсроченную постдеполяризацию, вследствие которой происходит разрыв деполяризующей волны [44]. Также активация NLRP3‑инфламмасомы в кардиомиоцитах усиливает сверхбыстрые K+ токи замедленного выпрямления (Ikur), тем самым уменьшая длительность и рефрактерность предсердного потенциала действия, что способствует поддержанию механизма re-entry [45][46]. Как было показано ранее, NLRP3‑инфламмасома может активироваться выходящими ионами калия, а значит, имеет место аутокринный путь регуляции запуска инфламмасомы K+. Действительно, было показано, что повышение экспрессии Kv1.5 каналов, функцией которых является индукция сверхбыстрого выхода ионов калия из кардиомиоцита, способствует активации NLRP3‑инфламмасомы [47]. Таким образом, активация инфламмасомы в кардиомиоцитах может вызывать электролитный дисбаланс клетки, сократительную дисфункцию и запускать аритмогенную активность. При ишемии миокарда в эндотелиальных клетках коронарных артерий повышалась экспрессия IL‑1β [48] и IL‑18 [27][28], что косвенно отражает активацию инфламмасомы в этих клетках.
Также известно, что в эндотелиоцитах при ишемической болезни сердца повышается экспрессия каспазы-1 [49]. Активация NLRP3‑инфламмасомы в эндотелиальных клетках коронарных артерий опосредует запуск воспалительных реакций и пироптоза самих эндотелиоцитов сосудов сердца [9]. Пироптоз эндотелиоцитов коронарных артерий может сопровождаться выходом компонентов инфламмасомы в межклеточное пространство, где они захватываются соседними эндотелиальными клетками, вызывая провоспалительные и атерогенные эффекты, которые могут быть обусловлены активацией NF‑κB и усилением экспрессии эндогенного NLRP3 в клетках-реципиентах без индукции их гибели [50]. Кроме этого, секреция клетками эндотелия коронарных артерий продуктов инфламмасомы приводит к привлечению и экстравазации лейкоцитов периферической крови, что способствует ремоделированию цитоскелета эндотелиальных клеток и увеличивает проницаемость сосудов, разрушая межклеточные контакты между соседними клетками [9].
Таким образом, активация различных видов инфламмасом в кардиомиоцитах, эндотелиальных клетках коронарных артерий и сердечных фибробластах приводит к ремоделированию сердечной ткани, нарушению ритма, образованию эктопических очагов, эндотелиальной дисфункции коронарных артерий, атерогенезу, а также развитию воспаления с дальнейшим формированием фиброза.
Отношения и деятельность. Исследование выполнено при финансовой поддержке Министерства науки и высшего образования Российской Федерации (соглашение № 075-15-2022-301 от 20.04.2022).
1. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36. doi:10.1038/s41421-020-0167-x.
2. Kelley N, Jeltema D, Duan Y, et al. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20(13):3328. doi:10.3390/ijms20133328.
3. Strowig T, Henao-Mejia J, Elinav E, et al. Inflammasomes in health and disease. Nature. 2012;481(7381):278-86. doi:10.1038/nature10759.
4. Ting JPY, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285-7. doi:10.1016/j.immuni.2008.02.005.
5. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229-65. doi:10.1146/annurev.immunol.021908.132715.
6. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6(1):291. doi:10.1038/s41392-021-00687-0.
7. Liu Z, Chen Y, Mei Y, et al. Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications. Molecules. 2023;28(23): 7813. doi:10.3390/molecules28237813.
8. Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. 2014;289(28):19571-84. doi:10.1074/jbc.M114.550624.
9. Bai B, Yang Y, Wang Q, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11(9):776. doi:10.1038/s41419-020-02985-x.
10. Bryan NB, Dorfleutner A, Kramer SJ, et al. Differential splicing of the apoptosisassociated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm Lond Engl. 2010;7:23. doi:10.1186/1476-9255-7-23.
11. Li Y, Fu TM, Lu A, et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A. 2018;115(43):10845-52. doi:10.1073/pnas.1810524115.
12. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi:10.1101/cshperspect.a008656.
13. Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193-206. doi:10.1016/j.cell.2014.02.008.
14. Van Opdenbosch N, Lamkanfi M. Caspases in Cell Death, Inflammation, and Disease. Immunity. 2019;50(6):1352-64. doi:10.1016/j.immuni.2019.05.020.
15. Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40(3):620-3. doi:10.1002/eji.200940185.
16. Lee DJ, Du F, Chen SW, et al. Regulation and function of the caspase-1 in an inflammatory microenvironment. J Invest Dermatol. 2015;135(8):2012. doi:10.1038/jid.2015.119.
17. Dai Y, Zhou J, Shi C. Inflammasome: structure, biological functions, and therapeutic targets. MedComm. 2023;4(5):e391. doi:10.1002/mco2.391.
18. Tang T, Gong T, Jiang W, et al. GPCRs in NLRP3 Inflammasome Activation, Regulation, and Therapeutics. Trends Pharmacol Sci. 2018;39(9):798-811. doi:10.1016/j.tips.2018.07.002.
19. He Y, Zeng MY, Yang D, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354-7. doi:10.1038/nature16959.
20. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002; 10(2):417-26. doi:10.1016/s1097-2765(02)00599-3.
21. He W ting, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285-98. doi:10.1038/cr.2015.139.
22. Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife. 4:e11205. doi:10.7554/eLife.11205.
23. Gaidt MM, Ebert TS, Chauhan D, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44(4):833-46. doi:10.1016/j.immuni.2016.01.012.
24. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519-50. doi:10.1146/annurev.immunol.021908.132612.
25. Sahoo M, Ceballos-Olvera I, del Barrio L, et al. Role of the Inflammasome, IL-1β, and IL-18 in Bacterial Infections. Sci World J. 2011;11:2037-50. doi:10.1100/2011/212680.
26. Chen G, Chelu MG, Dobrev D, et al. Cardiomyocyte Inflammasome Signaling in Cardiomyopathies and Atrial Fibrillation: Mechanisms and Potential Therapeutic Implications. Front Physiol. 2018;9:1115. doi:10.3389/fphys.2018.01115.
27. Pomerantz BJ, Reznikov LL, Harken AH, et al. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc Natl Acad Sci U S A. 2001;98(5):2871-6. doi:10.1073/pnas.041611398.
28. Toldo S, Mauro AG, Cutter Z, et al. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol - Heart Circ Physiol. 2018;315(6):H1553- H1568. doi:10.1152/ajpheart.00158.2018.
29. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol Baltim Md 1950. 2009;183(2):787-91. doi:10.4049/jimmunol.0901363.
30. Liao Y, Liu K, Zhu L. Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front Immunol. 2022;13:834289. doi:10.3389/fimmu.2022.834289.
31. Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28(5):886-92. doi:10.1248/bpb.28.886.
32. Yu L, Feng Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediators Inflamm. 2018;2018:9874109. doi:10.1155/2018/9874109.
33. Xu H, Yu W, Sun S, et al. TAX1BP1 protects against myocardial infarction-associated cardiac anomalies through inhibition of inflammasomes in a RNF34/MAVS/NLRP3- dependent manner. Sci Bull. 2021;66(16):1669-83. doi:10.1016/j.scib.2021.01.030.
34. Akosile W, Voisey J, Lawford B, et al. The inflammasome NLRP12 is associated with both depression and coronary artery disease in Vietnam veterans. Psychiatry Res. 2018;270: 775-9. doi:10.1016/j.psychres.2018.10.051.
35. Johansson Å, Eriksson N, Becker RC, et al. NLRC4 Inflammasome Is an Important Regulator of Interleukin-18 Levels in Patients With Acute Coronary Syndromes: GenomeWide Association Study in the PLATelet inhibition and patient Outcomes Trial (PLATO). Circ Cardiovasc Genet. 2015;8(3):498-506. doi:10.1161/CIRCGENETICS.114.000724.
36. Onódi Z, Ruppert M, Kucsera D, et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc Res. 2021;117(13):2639-51. doi:10.1093/cvr/cvab202.
37. Wang P, Zhang W, Feng Z, et al. LDL-induced NLRC3 inflammasome activation in cardiac fibroblasts contributes to cardiomyocytic dysfunction. Mol Med Rep. 2021;24(1):526. doi:10.3892/mmr.2021.12165.
38. Shi H, Gao Y, Dong Z, et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ Res. 2021;129(3):383-96. doi:10.1161/CIRCRESAHA.120.318629.
39. Artlett CM. The Mechanism and Regulation of the NLRP3 Inflammasome during Fibrosis. Biomolecules. 2022;12(5):634. doi:10.3390/biom12050634.
40. Artlett CM, Sassi-Gaha S, Rieger JL, et al. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011;63(11):3563-74. doi:10.1002/art.30568.
41. Artlett CM, Sassi-Gaha S, Hope JL, et al. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther. 2017;19(1):144. doi:10.1186/s13075-017-1331-z.
42. Yao C, Veleva T, Scott L, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138(20):2227-42. doi:10.1161/CIRCULATIONAHA.118.035202.
43. Heijman J, Muna AP, Veleva T, et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ Res. 2020;127(8):1036-55. doi:10.1161/CIRCRESAHA.120.316710.
44. Denham NC, Pearman CM, Caldwell JL, et al. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front Physiol. 2018;9:1380. doi:10.3389/fphys.2018.01380.
45. Gawałko M, Saljic A, Li N, et al. Adiposity-associated atrial fibrillation: molecular determinants, mechanisms, and clinical significance. Cardiovasc Res. 2023;119(3):614-30. doi:10.1093/cvr/cvac093.
46. Scott Jr L, Fender AC, Saljic A, et al. NLRP3 inflammasome is a key driver of obesityinduced atrial arrhythmias. Cardiovasc Res. 2021;117(7):1746-59. doi:10.1093/cvr/cvab024.
47. Li P, Kurata Y, Taufiq F, et al. Kv1.5 channel mediates monosodium urate-induced activation of NLRP3 inflammasome in macrophages and arrhythmogenic effects of urate on cardiomyocytes. Mol Biol Rep. 2022;49(7):5939-52. doi:10.1007/s11033-022-07378-1.
48. Galea J, Armstrong J, Gadsdon P, et al. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16(8):1000-6. doi:10.1161/01.atv.16.8.1000.
49. Zheng F, Gong Z, Xing S, et al. Overexpression of caspase-1 in aorta of patients with coronary atherosclerosis. Heart Lung Circ. 2014;23(11):1070-4. doi:10.1016/j.hlc.2014.04.256.
50. Gaul S, Schaeffer KM, Opitz L, et al. Extracellular NLRP3 inflammasome particles are internalized by human coronary artery smooth muscle cells and induce pro-atherogenic effects. Sci Rep. 2021;11(1):15156. doi:10.1038/s41598-021-94314-1.
Рубинштейн А. А. - лаборант-исследователь НИЛ аутоиммунных и аутовоспалительных заболеваний НЦМУ
Санкт-Петербург
Ходот А. А. - лаборант кафедры Факультетской терапии с клиникой
Санкт-Петербург
Тирикова П. В. - лаборант-исследователь НИЛ аутоиммунных и аутовоспалительных заболеваний НИЦ неизвестных, редких и генетически обусловленных заболеваний НЦМУ ЦПМ
Санкт-Петербург
Головкин А. С. - д.м.н., в.н.с., руководитель группы генно-клеточной инженерии, институт молекулярной биологии и генетики; в.н.с. НИЛ аутоиммунных и аутовоспалительных заболеваний НЦМУ
Санкт-Петербург
Кудрявцев И. В. - к.б.н., доцент, зав. лабораторией НИЛ аутоиммунных и аутовоспалительных заболеваний НЦМУ; зав. лабораторией клеточной иммунологии, ФГБНУ Институт экспериментальной медицины РАН
Санкт-Петербург
Шляхто Е. В. - д.м.н., профессор, академик РАН, генеральный директор
Санкт-Петербург
Рубинштейн А.А., Ходот А.А., Тирикова П.В., Головкин А.С., Кудрявцев И.В., Шляхто Е.В. Инфламмасома - новый взгляд на терапию сердечно-сосудистых заболеваний: обзор. Часть I. Российский кардиологический журнал. 2024;29(11S):5986. https://doi.org/10.15829/1560-4071-2024-5986
Rubinstein A.A., Khodot A.A., Tirikova P.V., Golovkin A.S., Kudryavtsev I.V., Shlyakhto E.V. Inflammasome - a new look at the therapy of cardiovascular diseases: a review. Part I. Russian Journal of Cardiology. 2024;29(11S):5986. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5986













































