


Содержание
Перейти к:
 С. Е. Андреева,
А. В. Гурщенков,
В. В. Зайцев,
А. А. Козырева,
А. И. Михалева,
А. С. Муравьев,
В. К. Гребенник,
Л. Б. Митрофанова,
М. Л. Гордеев,
О. М. Моисеева,
А. А. Костарева
С. Е. Андреева,
А. В. Гурщенков,
В. В. Зайцев,
А. А. Козырева,
А. И. Михалева,
А. С. Муравьев,
В. К. Гребенник,
Л. Б. Митрофанова,
М. Л. Гордеев,
О. М. Моисеева,
А. А. Костарева https://doi.org/10.15829/1560-4071-2024-5942
EDN: AIUPFZ
Перейти к:
Цель. Определить частоту и спектр редких вариантов гена филамина С (FLNC) среди пациентов с обструктивной гипертрофической кардиомиопатией (ГКМП), направленных на септальную миоэктомию, а также дать клиническую характеристику ГКМП, протекающей на фоне данных вариантов.
Материал и методы. 98 взрослым пациентам с ГКМП, которым была проведена процедура септальной миоэктомии, было выполнено генетическое тестирование методом секвенирования нового поколения с использованием целевой кардиопанели (панель на 39 генов применена у 58 пациентов, и панель на 17 генов — у 40 пациентов). У пациентов с редкими вариантами FLNC (с частотой минорного аллеля <0,01%) были проанализированы данные анамнеза, эхокардиографии (ЭхоКГ), электрокардиографии, холтеровского мониторирования, а также гистологического исследования миокарда, полученного интраоперационно.
Результаты. Выявлено 4 пациента с редкими вариантами FLNC (2 мужчины и 2 женщины), что составило 4% (Pro1774Ser, Thr1317Pro и His1834Tyr, последний выявлен дважды). Указанные варианты являлись точечными однонуклеотидными заменами и классифицировались как варианты неопределенной клинической значимости. Вариант FLNC p.Thr1317Pro у одного пациента сочетался с патогенным вариантом в гене MYH7 p.Val606Leu. Дебют заболевания у всех пациентов произошел после 40 лет. Клиническая картина была представлена умеренными проявлениями сердечной недостаточности и стенокардии на уровне II функционального класса, не было зарегистрировано пароксизмов неустойчивой желудочковой тахикардии и фибрилляции предсердий, клинически значимых блокад проводимости. При ЭхоКГ у одной пациентки с вариантом p.His1834Tyr наблюдалась двояковыпуклая межжелудочковая перегородка с преобладающей гипертрофией срединных отделов, в то время как у остальных пациентов была преимущественно гипертрофия базальных отделов. Диастолическая дисфункция не превышала 1-2 степени у всех четырех пациентов.
Заключение. Клинические характеристики носителей редких вариантов FLNC в нашем исследовании не отличались от большинства пациентов с ГКМП, подвергшихся операции септальной миоэктомии. Редкие варианты гена FLNC могут выступать в качестве причинных или модификаторов течения ГКМП. Для уточнения характера патогенности редких вариантов FLNC необходимы функциональные и популяционные исследования с применением сегрегационного анализа.
Андреева С.Е., Гурщенков А.В., Зайцев В.В., Козырева А.А., Михалева А.И., Муравьев А.С., Гребенник В.К., Митрофанова Л.Б., Гордеев М.Л., Моисеева О.М., Костарева А.А. Редкие варианты гена филамина С среди пациентов с гипертрофической кардиомиопатией, направленных на септальную миоэктомию. Российский кардиологический журнал. 2024;29(10):5942. https://doi.org/10.15829/1560-4071-2024-5942. EDN: AIUPFZ
Andreeva S.E., Gurshchenkov A.V., Zajcev V.V., Kozyreva A.A., Mihaleva A.I., Murav'ev A.S., Grebennik V.K., Mitrofanova L.B., Gordeev M.L., Moiseeva O.M., Kostareva A.A. Rare filamin C variants among patients with hypertrophic cardiomyopathy referred for septal myectomy. Russian Journal of Cardiology. 2024;29(10):5942. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5942. EDN: AIUPFZ
Гипертрофическая кардиомиопатия (ГКМП) является самым распространенным генетически обусловленным заболеванием сердца и характеризуется гипертрофией миокарда, неадекватной гемодинамической нагрузке [1][2]. Она ассоциирована с неблагоприятными исходами — фибрилляцией предсердий, сердечной недостаточностью (СН) и внезапной сердечной смертью [1][2]. В последние годы появляются новые данные, расширяющие представление об этиологии ГКМП, выводя ее за пределы "болезни саркомера": наряду с подтверждением полигенного вклада несаркомерных генов в развитие ГКМП, в спектре моногенных причин также появляются новые несаркомерные гены-кандидаты [3-5].
Примером является ген филамина С (FLNC), экспрессируемый в поперечно-полосатой мускулатуре [6][7]. В кардиомиоцитах FLNC локализован в области Z-диска, где он, соединяясь с тонкими филаментами, обеспечивает механическую стабильность саркомера [6][7]. Расположение в Z-диске и его взаимодействие с другими протеинами данной зоны позволяет ему выполнять функцию узла, обеспечивающего механосенсинг и трансдукцию сигнала [6]. FLNC — динамичный протеин, преобладающая локализация которого может меняться на различных этапах онтогенеза, а также в условиях стресса [6]. В субсарколемме FLNC является частью аппарата костамера, поддерживающего тонкие взаимодействия между саркомером и внеклеточным матриксом [6]. Также FLNC представлен в области вставочных дисков, где его количество повышается в условиях механического стресса [6].
Первые сообщения о выявлении патогенных вариантов FLNC относились к миопатиям, симптомы которых также могли включать гипертрофию левого желудочка [3, 8]. К настоящему моменту большинство сообщений о выявлении вариантов гена FLNC при заболеваниях относятся именно к кардиомиопатиям [3]. Укорачивающие варианты FLNC приводят к развитию дилатационной (ДКМП) и аритмогенной кардиомиопатии правого желудочка по механизму гаплонедостаточности, в то время как миссенс-варианты выявляются при ГКМП и рестриктивной кардиомиопатии (РКМП) и, как предполагается, формируют фенотип заболевания за счет неправильного фолдинга и агрегации протеинов [6][7][9][10]. Согласно исследованию Ader F, et al., варианты FLNC, которые можно идентифицировать как причинные, выявляются в 1,3% случаев ГКМП [10].
На настоящий момент остается актуальным вопрос о патогенности выявляемых у пациентов с ГКМП вариантов в гене FLNC, поскольку подавляющее их большинство являются миссенс и классифицируются как варианты неопределенной клинической значимости [3][7]. Изучение корреляции генотип-фенотип у пациентов с вариантами FLNC и ГКМП с более тяжелым клиническим течением может представлять актуальную научную задачу в рамках интерпретации вариантов FLNC при данной кардиомиопатии.
Наличие обструкции выносящего тракта левого желудочка (ВТЛЖ) является одним из ключевых эхокардиографических признаков, позволяющим идентифицировать подгруппу повышенного риска развития неблагоприятных событий [11][12]. Так, подавляющее большинство случаев СН при ГКМП формируется у пациентов с обструктивной формой, также наличие обструкции ВТЛЖ является фактором, повышающим риск развития внезапной сердечной смерти [11][12]. Пациенты с обструкцией ВТЛЖ имеют более выраженную диастолическую дисфункцию, выявляемую при эхокардиографии (ЭхоКГ), и более распространенный фиброз по данным магнитно-резонансной томографии, который локализован не только фокально в области гипертрофированного миокарда, но и носит диффузный характер, что, как предполагается, обусловлено хронической перегрузкой давлением [13]. Симптомным пациентам, рефрактерным к медикаментозной терапии, с тяжелой обструкцией ВТЛЖ, определяемой по пиковому градиенту в ВТЛЖ ≥50 мм рт.ст., экспертными сообществами рекомендовано проведение процедуры септальной миоэктомии [1][2].
На настоящий момент в литературе отсутствуют данные о спектре вариантов гена FLNC в когорте пациентов с обструктивной ГКМП и клинических характеристиках пациентов из данной генетической группы.
С помощью технологии секвенирования нового поколения, цель исследования — определить частоту и спектр редких вариантов FLNC среди взрослых пациентов с обструктивной ГКМП, направленных на септальную миоэктомию, дать клиническую и патоморфологическую характеристику ГКМП, протекающей на фоне данных вариантов.
Исследование было проведено в соответствии с Хельсинкской декларацией и одобрено локальным этическим комитетом НМИЦ им. В. А. Алмазова, перед включением у всех участников было получено письменное информированное согласие.
В исследование включено 98 неродственных пациентов с ГКМП, госпитализированных в НМИЦ им. В. А. Алмазова с целью проведения процедуры септальной миоэктомии (рис. 1). Для 58 пациентов было выполнено генетическое исследование методом секвенирования нового поколения с использованием целевой кардиопанели для проверки 39 генов, ассоциированных с развитием кардиомиопатий (список генов в Приложении). Еще 40 пациентам генетическое исследование методом массового параллельного секвенирования было выполнено с применением целевой панели на 17 генов (список генов в Приложении). Секвенирование библиотек было проведено с использованием набора целевого обогащения SureSelect Human All Exon V6 r2 (60Mbp) (Agilent Technologies, США) при помощи секвенатора Illumina HiSeq и SBSv4 реактивов (Illumina, США). Выравнивание, обработка данных и аннотации были выполнены с использованием референса генома человека hg19.
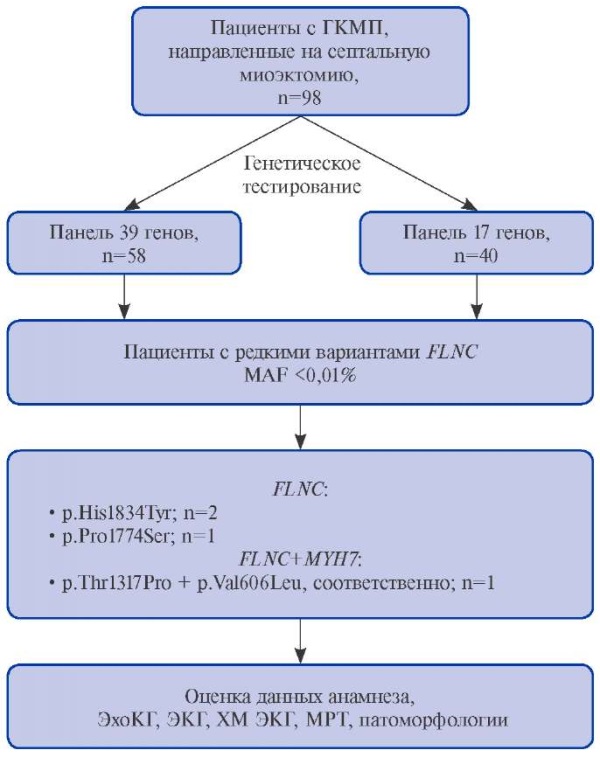
Рис. 1. Схема исследования.
Сокращения: ГКМП — гипертрофическая кардиомиопатия, МРТ — магнитно-резонансная томография, ХМ ЭКГ — холтеровское мониторирование ЭКГ, ЭКГ — электрокардиограмма, ЭхоКГ — эхокардиография, n — количество пациентов, MAF — частота минорного аллеля.
По результатам проведенного генетического тестирования были отобраны пациенты с редкими вариантами FLNC (MAF <0,01%), последние были классифицированы согласно рекомендациям Американской коллегии медицинской генетики (ACMG) [14]. Для отобранных пациентов были проанализированы данные анамнеза, ЭхоКГ, электрокардиограммы, выполненные до миоэктомии, холтеровского мониторирования. Дана патоморфологическая характеристика миокарда межжелудочковой перегородки (МЖП), полученного интраоперационно в ходе процедуры миоэктомии. Работа выполнена при финансовой поддержке гранта РНФ 15-20-00271 П.
Генетическая характеристика. Среди 98 проанализированных пациентов выявлено 4 пациента с редкими вариантами FLNC — 2 мужчины и 2 женщины (рис. 1, табл. 1). Данные варианты определены как миссенс и находились в гетерозиготном состоянии: p.Pro1774Ser, p.His1834Tyr и p.Thr1317Pro, при этом вариант p.His1834Tyr был обнаружен дважды, у двух неродственных пациентов (пациенты А-Г, соответственно). Все указанные варианты согласно ACMG/AMP классифицированы как варианты неопределенной клинической значимости. Ни один из указанных вариантов FLNC не был описан ранее в литературе. Вариант FLNC p.Thr1317Pro выявлен в сочетании с патогенным вариантом в гене MYH7 p.Val606Leu (табл. 1). Вариант FLNC p.Thr1317Pro был локализован в домене ROD1, остальные — в ROD2. Анализ предсказания патогенности in silico на основе вычислительных моделей продемонстрировал доказательства в пользу патогенного характера вариантов p.His1834Tyr и p.Thr1317Pro и доброкачественного характера для варианта p.Pro1774Ser (табл. 2).
Таблица 1
Клинические характеристики пациентов с вариантами FLNC
Пациент А | Пациент Б | Пациент В | Пациент Г | ||
Панель | 39 генов | 39 генов | 17 генов | 39 генов | |
Пол | М | Ж | Ж | М | |
Ген | FLNC | FLNC | FLNC | MYH7 | FLNC |
Вариант | p.Pro1774Ser | p.His1834Tyr | p.His1834Tyr | p.Val606Leu | p.Thr1317Pro |
Возраст дебюта заболевания | 45 | 41 | 64 | 43 | |
Возраст миоэктомии, лет | 49 | 42 | 66 | 45 | |
Жалобы | одышка при ФН, пресинкопы | одышка, боли в грудной клетке | одышка при ФН, боли в грудной клетке | одышка при ФН, синкопальные состояния | |
ХСН, ФК | II | II | II | II | |
Артериальная гипертензия | есть, контролируемая | нет | есть, контролируемая | нет | |
Сахарный диабет, тип 2 | нет | нет | нет | нет | |
Фибрилляция предсердий | нет | нет | нет | нет | |
Нарушения проводимости | не выявлено | блокада передне-верхнего разветвления ЛНПГ | не выявлено | блокада передне-верхнего разветвления ЛНПГ | |
Неустойчивая желудочковая тахикардия | нет | нет | нет | нет | |
Гемодинамически значимые стенозы КА | нет | интрамиокардиальное прохождение ПМЖА, стеноз в систолу 75% | нет | интрамиокардиальное прохождение ПМЖА, стеноз в систолу 70% | |
Максимальный градиент в покое, мм рт.ст. | 56 | 72 | 12 | 104 | |
Максимальный градиент провоцируемый, мм рт.ст. (метод провокации) | 132 (проба Вальсальвы) | 142 (проба Вальсальвы) | 65 (стресс-ЭхоКГ) | 126 (проба Вальсальвы) | |
Толщина МЖП, мм | 22 (базально) | 20 (базально) 29 (срединные отделы) | 19 (базально) | 20 (базально) | |
ЗС ЛЖ, мм | 13 | 12 | 14 | 10 | |
ФВ ЛЖ Симпсон, % | 66 | 60 | 57 | 77 | |
иОЛП, мл/м² | 47 | 45 | 39 | 41 | |
иКДО, мл/м² | 48 | 46 | 28 | 56 | |
иКСО, мл/м² | — | 19 | 12 | 13 | |
Митральная регургитация | умеренная | умеренная | легкая | умеренная | |
РСДЛА, мм рт.ст. | не лоцируется | 38 | 35 | 40 | |
Диастолическая дисфункция, степень | 2 | 2 | 1 | 2 | |
Сокращения: Ж — женский пол, ЗС ЛЖ — задняя стенка левого желудочка, иКДО — индексированный конечный диастолический объем, иКСО — индексированный конечный систолический объем, иОЛП — индекс объема левого предсердия, КА — коронарные артерии, ЛНПГ — левая ножка пучка Гиса, М — мужской пол, МЖП — межжелудочковая перегородка, ПМЖА — передняя межжелудочковая артерия, РСДЛА — расчетное систолическое давление в легочной артерии, ФВ ЛЖ — фракция выброса левого желудочка, ФК — функциональный класс, ФН — физическая нагрузка, ХСН — хроническая сердечная недостаточность, ЭхоКГ — эхокардиография.
Таблица 2
Редкие варианты гена FLNC, выявленные среди пациентов в данном исследовании, и их оценка по шкалам предсказания патогенности in silico
Пациенты | А | Б, В | Г |
Тип варианта | Миссенс | Миссенс | Миссенс |
Домен | ROD2 | ROD2 | ROD1 |
Экзон | 32 | 33 | 33 |
Позиция g. | g.128490459C>T | g.128490958C>T | g.128486202A>G |
Позиция c. | c.5320C>T | c.5500C>T | c.3949A>G |
Позиция p. | p.Pro1774Ser | p.His1834Tyr | p.Thr1317Pro |
Номер rs | rs370823820 | rs377141822 | rs377555574 |
MAF | 0.000007 | 0.00006083 | 0.00008034 |
ACMG/AMP | VUS | VUS | VUS |
In-silico predictors | Benign Supporting - BP4 | Pathogenic Supporting - PP3 | Pathogenic Supporting - PP3 |
MetaSVM | ND | Benign Moderate | Pathogenic Moderate |
FATHMM | Uncertain | Benign Supporting | Uncertain |
FATHMM-XF | Benign Supporting | Pathogenic Supporting | Pathogenic Moderate |
PROVEAN | Benign Supporting | Pathogenic Supporting | Pathogenic Supporting |
MutationTaster | Benign Supporting | Uncertain | Uncertain |
CADD | 23.1 | 26.7 | 25.4 |
Клиническая характеристика. У всех четырех пациентов заболевание дебютировало после 40 лет — в 41, 43, 45 и 64 года, — что потребовало проведения септальной миоэктомии спустя 1-4 года (табл. 1). Клиническая картина была представлена умеренными проявлениями СН и стенокардии на уровне II функционального класса; у пациента с вариантами FLNC+MYH7 наблюдались также синкопальные состояния. Не было зарегистрировано пароксизмов неустойчивой желудочковой тахикардии и фибрилляции предсердий. По данным проанализированных историй болезней не было указаний на наследственный характер заболевания.
Анализ ЭхоКГ и данных магнитно-резонансной томографии показал, что все пациенты с вариантами FLNC характеризовались промежуточной степенью выраженности гипертрофии левого желудочка без экстремальных значений: максимальная толщина стенок составила от 19 до 29 мм. У пациентки Б с вариантом p.His1834Tyr наблюдалась двояковыпуклая МЖП, что приводило к формированию обструкции на уровне базальных и срединных отделов, в то время как у остальных пациентов преобладал вариант с гипертрофией преимущественно базальных отделов МЖП (рис. 2). Ни один из пациентов на момент исследования не достиг гипокинетической фазы ГКМП, определяемой при фракции выброса по Симпсон <50%. У всех пациентов диастолическая дисфункция соответствовала 1 или 2 степени, дилатация левого предсердия также не превышала умеренных значений (индекс объема левого предсердия от 37 до 45 мл/м²), а показатели расчетного систолического давления в легочной артерии соответствовали легочной гипертензии 1 степени. Клинические характеристики пациентов с редкими вариантами FLNC суммированы в таблице 1.
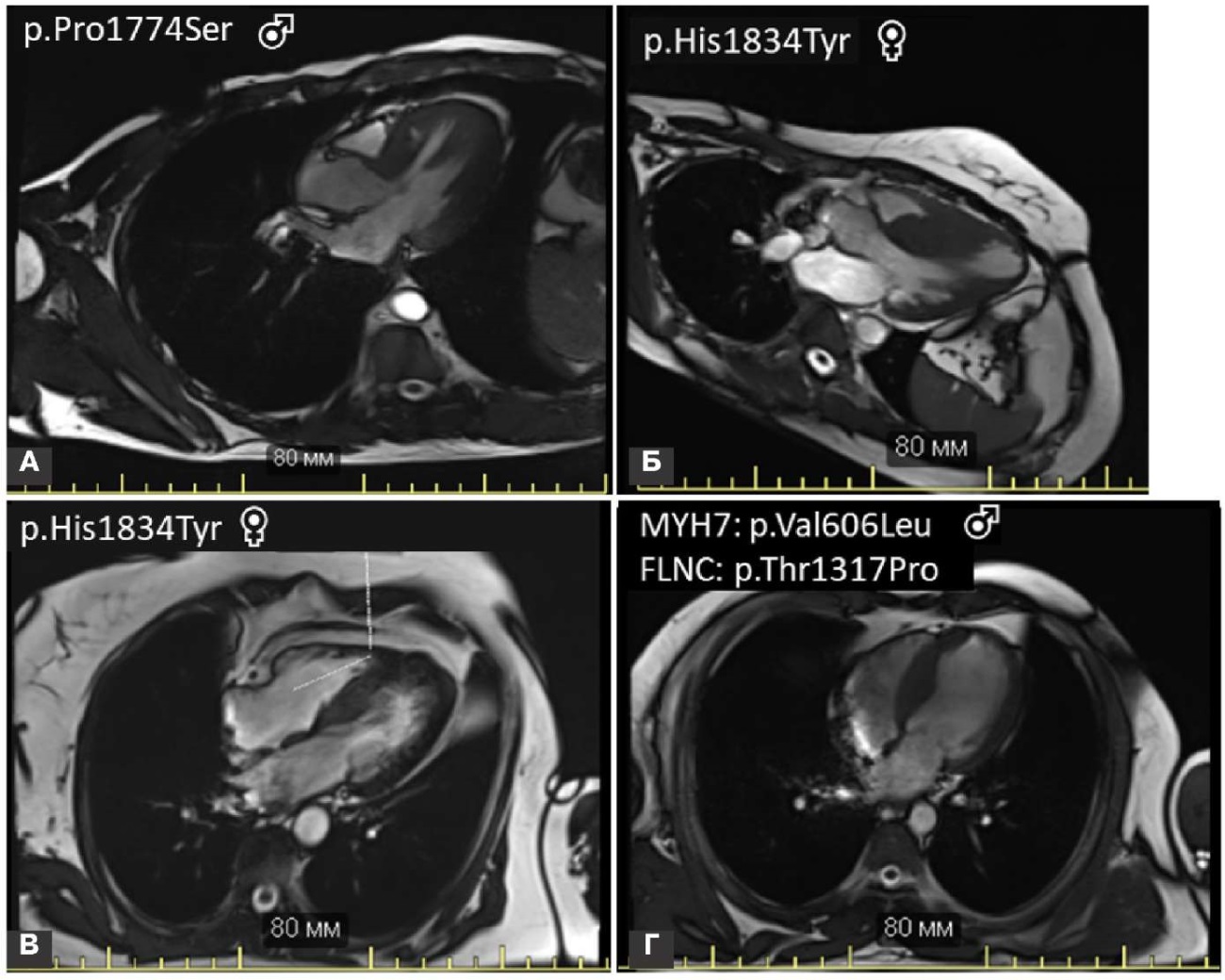
Рис. 2. Результаты магнитно-резонансной томографии носителей миссенс-вариантов FLNC. У пациентки Б с вариантом p.His1834Tyr наблюдалась двояковыпуклая МЖП с преобладающей гипертрофией на уровне срединных отделов, в то время как у остальных пациентов наблюдался вариант с гипертрофией преимущественно базальных отделов МЖП.
Во фрагментах миокарда МЖП, полученного интраоперационно, у всех пациентов наблюдались признаки ГКМП, такие как гипертрофия кардиомиоцитов, феномен "disarray" (дискомплексация) различной степени выраженности и мелкоочаговый фиброз (табл. 3). Дискомплексация кардиомиоцитов >15% выявлена у пациента с вариантом FLNC p.Pro1774Ser, а также у пациента с вариантами MYH7+FLNC p.Val606Leu и p.Thr1317Pro, соответственно, тогда как у оставшихся пациентов disarray составил <15%. Кроме того, у всех пациентов наблюдалось утолщение и фиброз эндокарда. Не было выявлено признаков амилоидоза и гликогеноза.
Таблица 3
Результаты патоморфологического исследования миокарда пациентов с вариантами FLNC среди миоэктомированных
Пациент | А | Б | В | Г |
Дискомплексация мышечных волокон | <15% | >15% | <15% | >15% |
Гипертрофия КМЦ | + | + | + | + |
Утолщение эндокарда | + | + | + | + |
Утолщение интрамуральных сосудов | + | + | — | — |
Реакция Шифф-йодной кислотой+включения | нет | есть (единичные КМЦ) | нет | нет |
Сокращение: КМЦ — кардиомиоциты.
В данной работе мы сфокусировались на вариантах гена FLNC, претендующего на роль причинного в развитии несаркомерных форм ГКМП. Особенность этого исследования заключается в том, что оно выполнено в когорте пациентов с обструктивной ГКМП, отобранных для проведения процедуры септальной миоэктомии, а значит, представляющих собой подгруппу повышенного риска развития неблагоприятных клинических исходов [11][12].
Изучение генетических основ и патогенетических механизмов формирования несаркомерной ГКМП имеет особый смысл на современном этапе лечения данной патологии: появление терапии обструктивной ГКМП ингибиторами сердечного миозина, основные механизмы работы которого изучены на клетках с мутациями генов саркомера, поднимает вопросы о потенциальной эффективности данной терапии в когорте пациентов без патогенных вариантов в генах саркомера, которая составляет 40-60% всех пациентов с ГКМП [4][5][15][16].
Среди 98 проанализированных пациентов редкие варианты FLNC были выявлены у 4 пациентов, что составляет 4%, согласуется с частотой редких вариантов FLNC в общей популяции пациентов с ГКМП, варьирующей по данным литературы от 1,3 до 5,0% [10][17-19]. Два из трех выявленных вариантов (p.Pro1774Ser и p.His1834Tyr) расположены в домене ROD2, являющемся мутационной горячей точкой [6][7]. Он является важным доменом для внутриклеточного сигналлинга, димеризации филамина и взаимодействия с Z-диском, в связи с чем, как предполагается, миссенс-варианты, локализованные в домене ROD2, имеют высокую вероятность быть патогенными для ГКМП [7]. Замена p.Thr1317Pro обнаружена в домене ROD1, являющемся еще одной распространенной локализацией вариантов для FLNC-ГКМП [6][7]. Ни один из указанных вариантов не был описан ранее в литературе.
Клиническое течение у носителей редких вариантов FLNC в нашем исследовании характеризовалось умеренной тяжестью: дебют заболевания произошел после 40 лет, СН не достигала продвинутых стадий, не было зафиксировано прогностически неблагоприятных желудочковых нарушений ритма и фибрилляции предсердий. Данные ЭхоКГ продемонстрировали легкую или умеренную степень дилатации левого предсердия, диастолическую дисфункцию первой или второй стадии, отсутствие снижения контрактильности и рестриктивного кровотока. Эти результаты также согласуются с данными литературы, где по основным клиническим характеристикам большинство носителей вариантов FLNC не отличались значимо от остальных пациентов с ГКМП [17][18].
К настоящему моменту выявлены определенные закономерности в реализации фенотипа FLNC-кардиомиопатий, в зависимости от функциональных характеристик причинного генетического варианта. Так, известно, что укорачивающие варианты FLNC (truncFLNC) преобладают у пациентов с ДКМП и аритмогенной дисплазии правого желудочка. Такие варианты несут в себе стоп-кодон и, как предполагается, запускают высококонсервативный механизм нонсенс-опосредованного распада РНК, в результате которого мутантный FLNC не синтезируется, количество FLNC в клетке снижается и возникает его гаплонедостаточность [6]. Данный механизм был заподозрен на основании того, что в цитоплазме кардиомиоцитов пациентов с truncFLNC-ДКМП не было выявлено агрегатов FLNC, в животных моделях truncFLNC с применением рыб данио-рерио и крысиных миобластов также не было выявлено агрегатов FLNC [6][9]. На гистологическом уровне у пациентов с truncFLNC-ДКМП наблюдаются участки выраженного циркулярного фиброза, что приводит к электрофизиологической нестабильности и прогностически неблагоприятным желудочковым аритмиям [9]. В связи с чем наличие truncFLNC у пациентов с ДКМП выделено отдельным пунктом в клинических рекомендациях при принятии решения об имплантации кардиовертера-дефибриллятора [1].
В свою очередь, у пациентов с ГКМП truncFLNC варианты никогда не были выявлены [20]. Вместо этого у пациентов с ГКМП и РКМП преобладают неукорачивающие варианты FLNC, из которых ~95% составляют миссенс-варианты [6]. Для миссенс-вариантов FLNC основным предполагаемым механизмом формирования фенотипа кардиомиопатии является неправильный фолдинг и агрегация протеинов, которые склонны локализовываться вокруг ядра [20-22]. С точки зрения клинических проявлений, большинство сообщений о РКМП на фоне миссенс-вариантов FLNC составляют случаи неблагоприятного течения с дебютом в детском возрасте, нередко в сочетании с нейромышечным фенотипом [21-26], в то время как ГКМП на фоне миссенс-вариантов FLNC не отличалась по своим клиническим характеристикам от общей когорты пациентов с ГКМП [17][18], с чем согласуются результаты нашего исследования. Исключения представляют собой единичные описания FLNC-ГКМП с яркой клинической картиной, неблагоприятным прогнозом и высокой семейной агрегацией [20][27].
По настоящий момент однозначная клиническая интерпретация редких вариантов FLNC, выявляемых среди пациентов с ГКМП, затруднена. Во-первых, для большинства выявляемых при ГКМП миссенс-вариантов FLNC отсутствуют функциональные исследования, в связи с чем они сохраняют за собой статус вариантов неопределенной клинической значимости. Во-вторых, имеются данные, ставящее под сомнение само положение о том, что варианты FLNC вызывают ГКМП: так, в работе Cui H, et al. 2018г частота вариантов FLNC в когорте 540 пациентов ГКМП не отличалась статистически значимо от таковой в группе 307 здоровых добровольцев [17], и, таким образом, могла отражать распространенность редких, но доброкачественных вариантов. Мы полагаем, что влияние миссенс-вариантов FLNC на реализацию фенотипа кардиомиопатий может представлять собой спектр: от модифицирующего влияния к вариантам с большего размера генетическим эффектом, что, вероятно, зависит от функциональных последствий конкретного генетического варианта.
Роль гистологических находок в диагностике и ведении ГКМП на современном этапе ее изучения остается неуточненной [28]. Характерными гистопатологическими признаками ГКМП являются гипертрофия и дискомплексация (феномен disarray) кардиомиоцитов, аномалии малых интрамуральных сосудов, интерстициальный и заместительный фиброз [28]. Известно, что феномен disarray более выражен у молодых пациентов с ГКМП, погибших от внезапной сердечной смерти, однако для установления четкой взаимосвязи между степенью дискомплексации и выживаемостью существующих данных недостаточно [28-30]. Заместительный фиброз связывают с ишемией, возникающей на фоне поражения малых сосудов, и он в большей степени, по-видимому, участвует в формировании осложнений при ГКМП, в то время как интерстициальный фиброз способствует диастолической дисфункции и дилатации предсердий [28][30]. В нашем исследовании у пациентов с вариантами FLNC при гистологическом исследовании фрагментов МЖП не было выявлено участков заместительного фиброза, но у всех был описан интерстициальный его вариант, что на клиническом уровне соответствовало умеренной или легкой диастолической дисфункции и отсутствию желудочковых нарушений ритма и, таким образом, согласуется с описанными выше литературными данными [28][30]. У всех пациентов с FLNC-ГКМП было выявлено утолщение эндокарда, характерное для пациентов старшей возрастной группы [30].
Отдельно мы бы хотели обсудить случай пациента Г, у которого было выявлено сочетание несаркомерного варианта FLNC p.Thr1317Pro неопределенной клинической значимости и патогенного варианта в гене саркомера MYH7 p.Val606Leu. Несмотря на то, что выявление патогенного саркомерного варианта само по себе достаточно для объяснения формирования фенотипа ГКМП, нам представляется важным сообщать о подобных клинических случаях в связи с доказанной неполной пенетрантностью саркомерных генетических вариантов и растущими свидетельствами полигенной природы ГКМП [3-5][31][32]. Сочетание вариантов FLNC с саркомерными описано в когорте пациентов с ГКМП: так, в исследовании Cui H, et al. у каждого третьего пациента с вариантом FLNC были также выявлены патогенные и вероятно патогенные варианты в генах саркомера [17]. Мы полагаем, что вариант FLNC p.Thr1317Pro также мог участвовать в формировании фенотипа заболевания, в роли модификатора или как самостоятельный причинный вариант.
К недостаткам нашего исследования следует отнести отсутствие сегрегационного анализа, а также отсутствие функциональных исследований для выявленных вариантов, в связи с чем их клиническая интерпретация затруднена, что требует дальнейшего экспериментального подтверждения и популяционных исследований.
В приведенных случаях клинические характеристики носителей редких миссенс-вариантов FLNC среди пациентов с обструктивной ГКМП, направленных на септальную миоэктомию, не отличались от большинства пациентов в общей когорте ГКМП и характеризовались дебютом заболевания после четвертой декады жизни, отсутствием гипокинетических форм, рестриктивного кровотока и жизнеугрожающих желудочковых аритмий. Дальнейшие функциональные и популяционные исследования должны пролить свет на корреляцию генотип-фенотип у пациентов с миссенс-вариантами FLNC и ГКМП.
Отношения и деятельность. Работа выполнена при финансовой поддержке гранта РНФ 15-20-00271 П.
1. Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur Heart J. 2023;44:3503-626. doi:10.1093/eurheartj/ehad194.
2. Габрусенко С. А., Гудкова А. Я., Козиолова Н. А. и др. Гипертрофическая кардиомиопатия. Клинические рекомендации 2020. Российский кардиологический журнал. 2021;26(5):4541. doi:10.15829/1560-4071-2021-4541.
3. Walsh R, Offerhaus JA, Tadros R, et al. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. 2022;19:151-67. doi:10.1038/S41569-021-00608-2.
4. Harper AR, Goel A, Grace C, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53:135-42. doi:10.1038/s41588-020-00764-0.
5. Tadros R, Francis C, Xu X, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53: 128-34. doi:10.1038/s41588-020-00762-2.
6. Song S, Shi A, Lian H, et al. Filamin C in cardiomyopathy: from physiological roles to DNA variants. Heart Fail Rev. 2022;27:1373-85. doi:10.1007/S10741-021-10172-Z.
7. Verdonschot JAJ, Vanhoutte EK, Claes GRF, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. 2020;41:1091-111. doi:10.1002/humu.24004.
8. Kley RA, Hellenbroich Y, Van Der Ven PFM, et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain. 2007;130 Pt 12:3250-64. doi:10.1093/BRAIN/AWM271.
9. Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016;68:2440-51. doi:10.1016/J.JACC.2016.09.927.
10. Ader F, De Groote P, Réant P, et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin Genet. 2019;96:317-29. doi:10.1111/CGE.13594.
11. Seferović PM, Polovina M, Bauersachs J, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2019;21:553-76. doi:10.1002/EJHF.1461.
12. O'Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35: 2010-20. doi:10.1093/EURHEARTJ/EHT439.
13. Avegliano G, Politi MT, Costabel JP, et al. Differences in the extent of fibrosis in obstructive and nonobstructive hypertrophic cardiomyopathy. J Cardiovasc Med. 2019;20:389-96. doi:10.2459/JCM.0000000000000800.
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. doi:10.1038/GIM.2015.30.
15. Kawana M, Spudich JA, Ruppel KM. Hypertrophic cardiomyopathy: Mutations to mechanisms to therapies. Front Physiol. 2022;13:975076.
16. Ho CY, Day SM, Ashley EA, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387-98. doi:10.1161/CIRCULATIONAHA.117.033200.
17. Cui H, Wang J, Zhang C, et al. Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy. Mol Genet genomic Med. 2018;6:1104-13. doi:10.1002/MGG3.488.
18. Gómez J, Lorca R, Reguero JR, et al. Screening of the Filamin C Gene in a Large Cohort of Hypertrophic Cardiomyopathy Patients. Circ Cardiovasc Genet. 2017;10:e001584. doi:10.1161/CIRCGENETICS.116.001584.
19. Thomson KL, Ormondroyd E, Harper AR, et al. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet Med. 2019;21:1576-84. doi:10.1038/S41436-018-0375-Z.
20. Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:article number 5326. doi:10.1038/NCOMMS6326.
21. Kiselev A, Vaz R, Knyazeva A, et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum Mutat. 2018;39:1161-72. doi:10.1002/HUMU.23559.
22. Brodehl A, Ferrier RA, Hamilton SJ, et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum Mutat. 2016;37:269-79. doi:10.1002/HUMU.22942.
23. Muravyev A, Vershinina T, Tesner P, et al. Rare clinical phenotype of filaminopathy presenting as restrictive cardiomyopathy and myopathy in childhood. Orphanet J Rare Dis. 2022;17. doi:10.1186/S13023-022-02477-5.
24. Schubert J, Tariq M, Geddes G, et al. Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy. Hum Mutat. 2018;39:2083-96. doi:10.1002/HUMU.23661.
25. Baban A, Alesi V, Magliozzi M, et al. Cardiovascular Involvement in Pediatric FLNC Variants: A Case Series of Fourteen Patients. J Cardiovasc Dev Dis. 2022;9. doi:10.3390/JCDD9100332.
26. Савостьянов К. В., Басаргина Е. Н., Рябова Е. Е. и др. Молекулярно-генетические особенности формирования рестриктивной кардиомиопатии у российских детей. Российский кардиологический журнал. 2021;26(10):4590. doi:10.15829/1560-4071-2021-4590.
27. Gaudreault N, Ruel LJ, Henry C, et al. Novel filamin C (FLNC) variant causes a severe form of familial mixed hypertrophic-restrictive cardiomyopathy. Am J Med Genet A. 2023;191:1508-17. doi:10.1002/AJMG.A.63169.
28. Rowin EJ, Fifer MA. Evaluating Histopathology to Improve Our Understanding of Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2021;77:2171-3. doi:10.1016/j.jacc.2021.03.292.
29. Varnava AM, Elliott PM, Mahon N, et al. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275-9. doi:10.1016/S0002-9149(01)01640-X.
30. Cui H, Schaff HV, Lentz Carvalho J, et al. Myocardial Histopathology in Patients With Obstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2021;77:2159-70. doi:10.1016/J.JACC.2021.03.008.
31. Lorenzini M, Norrish G, Field E, et al. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. J Am Coll Cardiol. 2020;76:550-9. doi:10.1016/J.JACC.2020.06.011.
32. Baulina NM, Kiselev IS, Chumakova OS, et al. Hypertrophic Cardiomyopathy as an Oligogenic Disease: Transcriptomic Arguments. Mol Biol (Mosk). 2020;54:955-67. doi:10.1134/S0026893320060023.
Андреева София Евгеньевна — врач-кардиолог, аспирант кафедры кардиологии, лаборант-исследователь.
Санкт-Петербург
Нет
Гурщенков Александр Викторович — к.м.н., доцент кафедры сердечно-сосудистой хирургии, врач сердечно-сосудистый хирург.
Санкт-Петербург
Нет
Зайцев Вадим Витальевич — врач-кардиолог, ассистент кафедры кардиологии факультета подготовки кадров высшей квалификации Института медицинского образования.
Санкт-Петербург
Нет
Козырева Александра Анатольевна — к.б.н., с.н.с. научно-исследовательской лаборатории молекулярной кардиологии и генетики.
Санкт-Петербург
Нет
Михалева Анна Игоревна — аспирант кафедры клеточной биологии, гистологии и цитологии.
Санкт-Петербург
Нет
Муравьев Алексей Сергеевич — врач детский кардиолог.
Санкт-Петербург
Нет
Гребенник Вадим Константинович — зав. отделением сердечно-сосудистой хирургии № 3.
Санкт-Петербург
Нет
Митрофанова Любовь Борисовна — д.м.н., г.н.с. НИЛ патоморфологии, профессор кафедры патологической анатомии Института медицинского образования.
Санкт-Петербург
Нет
Гордеев Михаил Леонидович — профессор, д.м.н., г.н.с. НИО кардиоторакальной хирургии.
Санкт-Петербург
Нет
Моисеева Ольга Михайловна — д.м.н., г.н.с., руководитель научно-исследовательского отдела некоронарогенных заболеваний сердца.
Санкт-Петербург
Нет
Костарева Анна Александровна — д.м.н., директор Института молекулярной биологии и генетики.
Санкт-Петербург
Нет
Андреева С.Е., Гурщенков А.В., Зайцев В.В., Козырева А.А., Михалева А.И., Муравьев А.С., Гребенник В.К., Митрофанова Л.Б., Гордеев М.Л., Моисеева О.М., Костарева А.А. Редкие варианты гена филамина С среди пациентов с гипертрофической кардиомиопатией, направленных на септальную миоэктомию. Российский кардиологический журнал. 2024;29(10):5942. https://doi.org/10.15829/1560-4071-2024-5942. EDN: AIUPFZ
Andreeva S.E., Gurshchenkov A.V., Zajcev V.V., Kozyreva A.A., Mihaleva A.I., Murav'ev A.S., Grebennik V.K., Mitrofanova L.B., Gordeev M.L., Moiseeva O.M., Kostareva A.A. Rare filamin C variants among patients with hypertrophic cardiomyopathy referred for septal myectomy. Russian Journal of Cardiology. 2024;29(10):5942. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5942. EDN: AIUPFZ













































