



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2024-5874
EDN: FQIHBM
Перейти к:
Среди различных типов дислипидемии семейная комбинированная гиперлипидемия (СКГЛ) является наиболее распространенным генетическим заболеванием, которое характеризуется, по крайней мере, двумя различными формами липидных нарушений: гиперхолестеринемией и гипертриглицеридемией. При наличии СКГЛ значительно повышается риск развития атеросклероз-ассоциированных сердечно-сосудистых заболеваний, в т. ч. в молодом возрасте. Цель работы — выполнить анализ литературных данных о современных критериях диагностики, патогенезе и данных молекулярно-генетических исследований СКГЛ. Будущие исследования, направленные на изучение лежащих в основе СКГЛ генетических и метаболических механизмов и разработку эффективных стратегий лечения, должны включать более крупные когортные исследования с более широким наследственным разнообразием, а также исследование эпигенетических факторов и факторов образа жизни.
Тимощенко О.В., Шахтшнейдер Е.В. Семейная комбинированная гиперлипидемия, современное состояние проблемы (обзор литературы). Российский кардиологический журнал. 2024;29(8):5874. https://doi.org/10.15829/1560-4071-2024-5874. EDN: FQIHBM
Timoshchenko O.V., Shakhtshneider E.V. Familial combined hyperlipidemia: current status of the problem (literature review). Russian Journal of Cardiology. 2024;29(8):5874. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5874. EDN: FQIHBM
Нарушения липидного обмена — дислипидемии (ДЛП) характеризуются изменением концентрации в плазме крови различных липидных и липопротеиновых фракций (общего холестерина (ОХС) и холестерина липопротеинов низкой плотности (ХС-ЛНП), холестерина липопротеинов очень низкой плотности (ХС-ЛОНП), триглицеридов (ТГ), хиломикронов). Нарушения обмена липидов вследствие генетических дефектов классифицируются как первичные. Первичные ДЛП включают гетерогенный набор моногенных и полигенных состояний, которым свойственны семейная агрегация, выраженная гиперхолестеринемия и/или гипертриглицеридемия, проявление в раннем возрасте и высокий риск сердечно-сосудистых событий и/или рецидива панкреатита [1].
Среди различных типов ДЛП семейная комбинированная гиперлипидемия (СКГЛ) (Е78.4 по классификации МКБ-10) наряду с семейной гиперхолестеринемией (Е78.0 по классификации МКБ-10) является широко распространенным генетическим заболеванием. В литературе СКГЛ также может встречаться под названием гиперлипопротеинемия или смешанная гиперлипидемия. Распространенность СКГЛ по различным данным составляет 1:50-1:200 человек. Так, ~3,5 млн пациентов с СКГЛ насчитывается в Европе, в Соединенных Штатах — 2,7 млн человек [1][2]. Trinder M, et al. (2022) провели ретроспективную оценку распространенности СКГЛ в Великобритании, используя различные версии диагностических критериев. Частота фенотипа СКГЛ согласно критериям Консенсусной конференции, Голландским, Мексиканским, Брунцелла и Гольдштейна составила, соответственно, 11,44% (n=39961), 5,01% (n=17485), 1,48% (n=5153), 1,10% (n=3838) и 0,48% (n=1688), имея значительную вариабельность [3]. В российской популяции данных о численности населения с СКГЛ нами не обнаружено. Кроме того, найдена лишь единичная публикация с описанием клинического наблюдения семьи с СКГЛ [4].
Цель работы — выполнить анализ литературных данных о современных критериях диагностики, патогенеза и данных молекулярно-генетических исследований СКГЛ.
Осуществлен поиск литературы в базе Pubmed. Схема представлена на рисунке 1. Работа выполнена в рамках темы Государственного задания № FWNR-2022-0003.

Рис. 1. Блок-схема отбора публикаций в PubMed для обзора.
Клинические проявления СКГЛ
В клинической картине у пациентов обычно наблюдается повышение уровней ХС-ЛОНП (IV тип по классификации Фридрексон), ХС-ЛНП (IIa тип) или их сочетание (IIb тип) ≥90-го процентиля для их возраста и пола с повышенным уровнем аполипопротеина (апо) В в качестве основного объединяющего субфенотипа [5]. При физикальном обследовании пациентов с СКГЛ нет специфических проявлений, но кожные ксантомы могут иногда встречаться при высоких уровнях ОХС или ТГ. У пациентов могут наблюдаться признаки сердечно-сосудистых заболеваний (ССЗ), таких как артериальная гипертония, заболевания периферических артерий, инфаркт миокарда или ишемический инсульт в анамнезе [6].
Определение клинических критериев СКГЛ неоднократно менялось с момента первоначальной характеристики заболевания [7], что затрудняет сравнение результатов различных исследований [8][9]. Некоторые критерии требуют наличия как минимум двух родственников первой степени родства с аномальным липидным фенотипом [7-10], тогда как другие определения отличаются точными предельными значениями повышенного уровня ХС-ЛНП и ТГ. Различия российских и европейских диагностических критериев СКГЛ представлены в таблице 1.
Таблица 1
Российские и европейские диагностические критерии СКГЛ
Параметр | Диагностические критерии СКГЛ | |
Российские [1] | Европейские [2] | |
апоB | >120 мг/дл | >120 мг/дл |
ТГ | >1,5 ммоль/л (>133 мг/дл) | >150 мг/дл |
Семейная история раннего дебюта ССЗ | + | - |
ХС-ЛВП | - | <1,0/1,2 ммоль/л |
Наличие мелких плотных частиц ЛНП | - | + |
Сокращения: апо — аполипопротеин, ЛНП — липопротеины низкой плотности, СКГЛ — семейная комбинированная гиперлипидемия, ССЗ — сердечно-сосудистые заболевания, ТГ — триглицериды, ХС-ЛВП — холестерин липопротеинов высокой плотности.
Сложность диагностики заболевания усугубляется тем, что СКГЛ имеет высокую вариабельность фенотипа у одного и того же человека с течением времени и в одной семье, что обусловливает низкую выявляемость, несмотря на высокий риск ССЗ. Также затруднение в постановке диагноза связано с коморбидностью СКГЛ с другими метаболическими заболеваниями, такими как ожирение, инсулинорезистентность, сахарный диабет 2 типа (СД2), гипертония, неалкогольная жировая болезнь печени и метаболический синдром [1][6][11-14]. При таких состояниях отмечаются более высокие уровни апоВ в крови по сравнению с наличием только состояния инсулинорезистентности. Кроме того, пациенты с СКГЛ имеют большую предрасположенность к развитию СД2 по сравнению с пациентами без наследственной ДЛП [6].
Интересны предложения исследователей способов определения ХС-ЛНП в сыворотке крови, поскольку ДЛП при СКГЛ характеризуется преобладанием богатых ТГ ЛОНП и несоответствием между уровнями ХС-ЛНП и апoB [15]. Ученые предлагают уравнения альтернативные формуле Фридвальда для более точной оценки уровня ХС-ЛНП при СКГЛ. Авторский метод Martin SS (2018) предполагает оценку ХС-ЛНП с помощью следующей формулы: ХС-ЛНП (мг/дл) = (ОХС—ХС-ЛВП) — (ТГ/коэффициент). В отличие от формулы Фридвальда, вместо деления ТГ на фиксированный коэффициент для ХС-ЛОНП, равный 5, в уравнение Martin в формулу введен регулируемый коэффициент для соотношения ТГ/ХС-ЛОНП, который сопоставляет ТГ каждого пациента и холестерина липопротеинов не-высокой плотности в 180 измерениях в диапазоне от 3,1 до 9,5, чтобы получить персональную оценку ЛОНП в мг/дл [16]. Другие исследователи Zubirán R, et al. (2023) представляют формулу Сэмпсона (S-LDL-C) и новое уравнение Сэмпсона (eS-VLDL-C), включающее апоB, которые показывают наименьшую погрешность, лучшие результаты по ТГ и ХС-ЛНП [17].
Ввиду того, что при СКГЛ увеличивается количество свободных жирных кислот в крови и приводит к длительному воздействию повышенного уровня апоВ, входящего в состав ХС-ЛНП и ХС-ЛОНП, значительно повышается риск развития атеросклероз-ассоциированных ССЗ, в т. ч. в молодом возрасте [18][19]. Распространенность ишемической болезни сердца (ИБС) у пациентов с СКГЛ моложе 60 лет составляет ~15% [20][21]. По данным канадских ученых пациенты с фенотипом СКГЛ имеют аналогичный риск развития ИБС по сравнению с участниками с моногенной семейной гиперхолестеринемией (скорректированное отношение рисков к контролю (95% доверительный интервал (ДИ)): 2,72 (2,31-3,21) и 1,90 (1,30-2,78)), несмотря на то, что встречается примерно в 5 раз чаще [3]. Luijten J, et al. (2019) оценили риск развития ССЗ (ИБС, ишемический инсульт и заболевания периферических артерий, требующие инвазивного лечения) у пациентов с СКГЛ, их супругов и родственников с нормальными показателями липидов (n=596). Медиана наблюдения составила 15 лет. Частота ССЗ была значительно выше у пациентов с СКГЛ, чем у их супругов (23,6% vs 4,7%; отношение рисков: 5,4, 95% ДИ: 2,0-14,6; отношение рисков после поправки на факторы риска, включенные в SCORE: 4,7, 95% ДИ: 1,6-13,8), одновременно различий в частоте ССЗ между группой родственников без ДЛП и группой супругов не выявлено (5,8% vs 4,7%) [22].
Молекулярно-генетические исследования СКГЛ
Впервые независимо друг от друга СКГЛ в разных когортах одновременно описали Goldstein JL, et al. в 1973 г, проанализировав характер ДЛП среди 2500 пациентов с инфарктом миокарда в анамнезе [7], и Kwiterovich PO, et al. [23]. Теория моногенной этиологии заболевания была основана на объединении фенотипических признаков и математическом моделировании, без выполнения анализа ДНК. Считалось, что наследование комбинированной гиперлипидемии лучше всего объясняется менделевским аутосомно-доминантным типом наследования, с уточнением, что экспрессия предполагаемого гена вариабельна [7]. Brahm AJ, et al. (2016), используя математическое моделирование, показали, что доля членов семьи с ДЛП больше, чем можно было бы ожидать при полигенном заболевании, и более свойственна доле пациентов, которая ожидается при моногенном заболевании. Кроме того, в модели предполагалось, что одновременное повышение уровня ТГ и ОХС объясняется одним и тем же геном, а не двумя отдельными, без значительной роли факторов окружающей среды [24].
Однако позже было обнаружено, что она может быть семейной или несемейной, что обусловлено моногенным или полигенным характером наследования [5, 20, 21, 25]. Несмотря на полувековой стаж молекулярно-генетических исследований данного заболевания, генетические аспекты до конца не изучены. Результаты большинства исследований однозначно показывают, что многочисленные однонуклеотидные варианты (ОНВ), расположенные в десятках позиций по всему геному, каждый из которых оказывает лишь незначительное или умеренное влияние на липиды, типичны для генетического профиля многих пациентов с СКГЛ [8, 20, 21, 24, 26-28]. Одно из крупных генетических исследований проведено финскими учеными Ripatti P, et al. (2016) [29], которые проанализировали 9 млн вариантов у 715 членов семей с ДЛП. Более трети (35%) пациентов имели либо повышенный уровень ТГ (25% пациентов), либо повышенный уровень ХС-ЛНП (17% пациентов), либо и то, и другое (6,8% пациентов). Авторы сообщают об увеличении частоты ОНВ, предрасполагающих либо к высокому уровню ХС-ЛНП, либо ТГ, и снижении частоты ОНВ, связанных с более низким уровнем ХС-ЛНП, в 3% обнаружены редкие патогенные варианты. В другой работе Gill PK, et al. (2021) [30] у пациентов комбинированной ДЛП использовали целевую панель секвенирования следующего поколения для определения полигенного риска повышения уровней ТГ и ХС-ЛНП, включающую 16 ОНВ. У пациентов с СКГЛ, как и у пациентов с изолированной гипертриглицеридемией, были значительно увеличены шансы на высокий полигенный балл по риску гипертриглицеридемии: 2,50 (95% ДИ: 1,61-3,88; р<0,001) и 3,72 (95% ДИ: 2,24-6,19; р<0,001), соответственно. У пациентов с СКГЛ не обнаружено значительного накопления редких вариантов, ассоциированных с повышением ХС-ЛНП или ТГ, и высокого полигенного показателя ХС-ЛНП. Taghizadeh E, et al. сообщают об участии, по крайней мере, 35 различных генетических вариантов в развитии СКГЛ (табл. 2) [20][21]. Редкие варианты с большим эффектом в основных генах липидного обмена были описаны в нескольких случаях СКГЛ [8][21][25][27-29], причем один и тот же редкий вариант иногда обнаруживается у нескольких членов с ДЛП одной семьи [21]. Однако ни редкие варианты в генах классического метаболизма ТГ (LPL, LMF1, APOA5, APOC2, GPIHBP1), ни в генах метаболизма ХС-ЛНП (LDLR, APOB, PCSK9), вероятно, не представлены в большем количестве при СКГЛ [30].
Таблица 2
Некоторые гены, ассоциированные с развитием СКГЛ (адаптировано из [25])
Ген | Локализация | Продукт | Оказывает воздействие | Функция | Вариант |
LIPE | 19q13 | гормон-чувствительная липаза | жировая ткань | липолиз | g.60C >G |
PNPLA2 | 11p15 | жировая триглицеридлипаза | жировая ткань | разрушает ТГ | |
GPR77 | 19q13 | белок, стимулирующий ацилирование | жировая ткань | липогенный гормон | Ser323Ile |
LEPR | 1p31.3 | рецептор лептина | жировая ткань | жировой обмен | 223 A/G |
PPARs | 3p253 | PPARα,β,γ ферменты | жировая ткань | регулирует триглицеридлипазу | Pro12Ala C161T |
USF1 | 1q21.23 | транскрипционный фактор | жировая ткань | регулирует транскрипцию генов | rs3737787 |
GCKR | 2p23 | регуляторный белок глюкокиназы | ХС-ЛОНП | регулятор глюкокиназы | Pro446Leu |
Apo E | 19q13 | лиганд | ХС | лиганд для рецептора апоE и ЛНП | p.Leu149del |
OSBPL10 | 3p22 | оксистеролсвязывающий белок | ХС | стероловый сенсор и регулятор процесса дефосфорилирования | rs11716163 |
LPL | 8p22 | LPL фермент | ТГ | катаболизм ТГ | p.Asp277Asn |
CETP | 16q12 | белок-переносчик эфиров холестерина | ХС-ЛВП | транспортер эфира холестерина между липопротеинами | rs173539 |
GALNT2 | 1q41.4 | полипептид N ацетилгалакт | ОХС | О-связанное гликозилирование | rs4846913 |
LCAT | 16q21 | лецитин-холестерин-ацилтрансфераза | ОХС | обратный транспорт холестерина | rs2271293 |
LIPC | 15q21 | триглицеридлипаза печени | ОХС | гидролиз ТГ и фосфолипидов | rs28933094 |
RXRγ | 1q21 | рецептор ретиноида X | ХС-ЛНП | транскрипционный фактор | p.Gly14Ser |
ANGPTL3 | 1p31.1 | ангиопоэтиноподобные секреторные белки | ХС-ЛОНП, ХС-ЛНП, ХС-ЛВП | связывается с липопротеинлипазой и подавляет ее функцию | S17X |
GPHDLBP1 | — | гликозилфосфатидилинозитолсвязывающий | хиломикроны | липопротеинлипаза порт и транспортер | c. (− 83G >A) |
LMF1 | 16 | фактор зрелости липазы 1 | ТГ | липаза-шаперон | Y439X |
LDLR | 19p13 | рецептор ЛНП | ХС-ЛНП | обнаруживает частицы ЛНП | — |
Apo B | 4q32.3 | апо В | ХС-ЛОНП | rs6829588 | |
PCSK9 | 1p32 | пропротеин-конвераза-субтилизин-кексин типа 9 | гепатоциты | гомеостаз холестерина | rs2479409 |
ATF6 | 1q22 | транскрипционный фактор | эндоплазматическая сеть | регулируют гомеостаз холестерина | Met Val |
ADD1 | 4p16 | аддуцин-1 | — | участвует в структуре цитоскелета | Gly460Trp |
APO-BEC1 | 12p13 | каталитический полипептид 1 | тонкая кишка | участвует в процессинге мРНК апоВ | rs1349411 |
CRABP2 | 12q21 | клеточный белок 2, связывающий ретиноевую кислоту | — | транскрипционный фактор | — |
FADS3 | 11q12 | десатураза жирных кислот 3 | жировая ткань | регулируют десатурацию жирных кислот | rs174547 |
FOXC2 | 16q24 | транскрипционный фактор Foxc2 | жировая ткань | способствует развитию лимфатической и сердечно-сосудистой систем | — |
GAL | 11q13 | препропептид галанин | ТГ | нейропептид | rs2187331 |
HNF4A | 20q13 | ядерный фактор гепатоцитов 4 | ХС-ЛНП | регулирует уровень глюкозы и липидов в сыворотке крови | rs1800961 |
CERS4 | 19p13.2 | керамидсинтаза | ХС-ЛВП | синтез сфинголипидов | rs17159388 |
PCDH15 | 10q21 | протокадгерин-15 | ТГ, апоВ, ОХС | опосредует кальций-зависимую межклеточную адгезию | rs10825269 |
PON1 | 7q21.3 | параоксоназы 1 | ХС-ЛВП | кальций-зависимая эстераза | Q192R Glu192Arg |
TCF7L2 | 10q25 | фактор, специфичный для Т-клеток | ТГ, ХС-ЛНП | сигнальный путь Wnt | rs7903146 rs12255372 |
TNFRSF1B | 1p36 | рецептор ФНО | Жировая ткань | неоваскуляризация | rs1061622 |
WWOX | 16q23.2 | WW-домен, содержащий оксидоредуктазу | ТГ, ХС-ЛВП | метаболизм стероидов | rs2059238 |
HMGCR | 5 | ГМГ-КоА-редуктаза | ХС-ЛНП | — | rs3846662 I638V |
Сокращения: апо — аполипопротеин, ЛНП — липопротеины низкой плотности, ОХС — общий холестерин, ТГ — триглицериды, ФНО — фактор некроза опухоли, ХС — холестерин, ХС-ЛВП — холестерин липопротеинов высокой плотности, ХС-ЛНП — холестерин липопротеинов низкой плотности, ХС-ЛОНП — холестерин липопротеинов очень низкой плотности.
Взаимодействие генетических вариантов с большим эффектом, кумуляция генетических вариантов с малым эффектом и триггеров окружающей среды способствует развитию фенотипа СКГЛ (рис. 2) [8].
В патофизиологические механизмы развития данного полигенного заболевания вовлечен комплекс метаболических путей, что приводит к гиперсекреции печенью содержащих апоВ липопротеинов в сочетании с замедленным выведением ХС-ЛОНП и остатков хиломикронов. Эти дефекты связаны с нарушением метаболизма свободных жирных кислот и резистентностью к инсулину на уровнях жировой, печеночной и мышечной тканей (рис. 3) [20].

Рис. 2. Кумулятивный вклад генетических вариантов с большим эффектом, генетических вариантов с малым эффектом и триггеров окружающей среды в развитии СКГЛ (адаптировано из [8]).
Сокращения: апо — аполипопротеин, ОХС — общий холестерин, СКГЛ — семейная комбинированная гиперлипидемия, ТГ — триглицериды, ХС-ЛНП — холестерин липопротеинов низкой плотности.
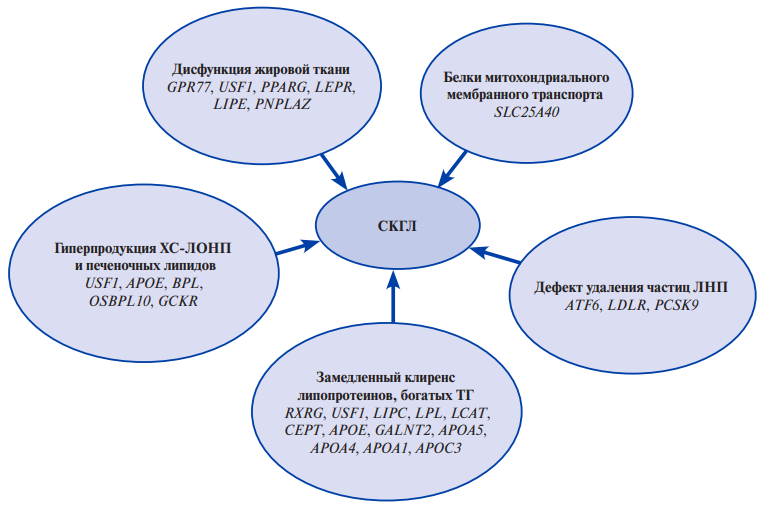
Рис. 3. Метаболические пути и ассоциированные с ними гены, участвующие в развитии СКГЛ (адаптировано из [20]).
Сокращения: ЛНП — липопротеины низкой плотности, СКГЛ — семейная комбинированная гиперлипидемия, ТГ — триглицериды, ХС-ЛОНП — холестерин липопротеинов очень низкой плотности.
Считается, что основным патофизиологическим механизмом при СКГЛ является избыточная продукция печенью липопротеиновых частиц, содержащих апоB-100, а именно ХС-ЛОНП и ХС-ЛНП, в виде дисбаланса между липогенезом de novo и β-окислением ассоциированным с инсулинорезистентностью, снижением скорости клиренса апоВ и увеличением экспрессии молекул, которые подавляют рецептор ЛНП. Это приводит к повышению уровня ОХС, ТГ и апоB. Кроме того, у лиц с СКГЛ снижен уровень ХС-ЛВП и увеличено количество мелких плотных ЛНП и остаточных липопротеиновых частиц [6][31]. Дисфункция жировой ткани характеризуется увеличением уровня свободных жирных кислот и оттоком их в печень, что приводит к увеличению скорости синтеза липопротеинов [6]. Существует тесная связь между синтезом холестерина в печени и ожирением, а избыточный синтез холестерина является одним из механизмов ДЛП при метаболическом синдроме и СД2 [32]. Исследование Baila-Rueda L, et al. (2018) также показывает, что для СКГЛ характерна более низкая абсорбция холестерина в кишечнике и его более высокий синтез независимо от возраста, пола, апоE и индекса массы тела по сравнению с первичной гиперхолестеринемией [33]. Известно, что повышенные уровни и продукция апоС-II и апоС-III являются детерминантами кинетики и концентрации в плазме липопротеинов, богатых ТГ, включая ХС-ЛОНП. Ген APOCIII также связан с состояниями инсулинорезистентности и СД2, которые часто ассоциируются с СКГЛ [34].
Несмотря на то, что роль гена USF1, кодирующего вышестоящий связывающий транскрипционный фактор 1, регулирующего экспрессию нескольких генов, участвующих в метаболизме липидов, глюкозы и жировой ткани, в патогенезе СКГЛ в настоящее время до конца не объяснена, он считается одним из генов-кандидатов, наиболее часто ассоциированным с этим типом ДЛП [8][35]. Около 20 лет назад Pajukanta P, et al. (2004) показали, что СКГЛ связана с общим гаплотипом, содержащим некодирующие ОНВ в гене USF1 [36]. В экспериментальных исследованиях на животных моделях Laurila PP, et al. (2016) определили, что инактивация USF1 оказывает протективный эффект в отношении ДЛП на фоне нарушения диеты, ожирения, стеатогепатоза и атеросклероза. Кроме того, в этой группе наблюдалась повышенная чувствительность к инсулину и снижение стеатоза печени по сравнению с мышами типа USF1+/+. Предложенный механизм связан с повышенным поглощением ТГ бурой жировой тканью посредством LPL-зависимого механизма, который усиливает адренергический ответ и термогенез в коричневых адипоцитах. В клиническом исследовании продемонстрированы схожие физиологические эффекты при снижении экспрессии мРНК USF-1, в виде улучшения чувствительности к инсулину, липидного профиля и замедления атерогенеза [37]. Ген USF1 в сочетании с транскрипционным фактором USF2 регулирует транскрипцию ~40 генов, в т. ч. аполипопротеинов, ферментов и связанных транспортеров. Данный механизм осуществляется путем связывания с элементом e-box выше гена APOA5, что усиливает его транскрипцию. В присутствии инсулина димер USF1 фосфорилируется, теряет аффинность связывания с e-box и, таким образом, снижает транскрипцию. Вполне вероятно, что несколько вариантов в этих генах, взаимодействующих между собой, могут привести к появлению сложного фенотипа СКГЛ [8][21][24][38]. Отсутствие патогенных вариантов в кодирующей области USF1 и низкая транскрипция у пациентов с СКГЛ усложняет исследования по выявлению молекулярных изменений, приводящих к такому сложному фенотипу. Изучение профиля транскрипции в биопсии жировых подушечек носителей определенных аллелей риска ОНВ USF1 указывает на значительные изменения в экспрессии некоторых генов, связанных с USF1, в виде снижения экспрессии APOE (приводящее к гипертриглицеридемии) и ABCA1 (приводящее к гипопаратиреозу), увеличение экспрессии ангиотензиногена (приводящее к гипертензии), а также APOCII, APOAII, печеночной липазы, глюкокиназы [35]. Данные проявления коррелируют с фенотипом СКГЛ, что позволяет предположить, что изменение USF1 может оказывать значимое влияние на гены-мишени, связанные с молекулярным патогенезом ДЛП [8][21][24][38]. На сегодняшний день идентифицировано >3,5 тыс. вариантов в гене USF1 по данным базы dbSNP (https://www.ncbi.nlm.nih.gov/snp/?term=USF1). Интерес к USF1 сохраняется в настоящее время, и Taghizadeh E, et al. (2019) определили новый патогенный вариант основателя p.Arg196Trp в USF1 у всех 13 членов с СКГЛ из одной семьи, при этом он отсутствовал у всех незатронутых членов семьи и контрольной группы, что предполагает неслучайную статистическую связь генотип-фенотип [35]. Ранее в других исследованиях были предложены механизмы патогенности вариантов в USF1, включая недостаточную транскрипцию нижестоящих генов, регулирующих окисление жирных кислот в жировой ткани [39], а также обратная связь между USF1 и FOXA2, которая влияет на печеночную секрецию ТГ [40].
APOE является еще одним основным геном, который, как предполагалось ранее, вносит вклад в фенотип СКГЛ, его экспрессия частично регулируется USF1 [35][41]. Однако единого мнения о связи APOE с СКГЛ в настоящее время нет. Генотип ɛ2/ɛ2, предрасполагает к развитию дисбеталипопротеинемии, которая отличается от СКГЛ и характеризуется накоплением остаточных липопротеиновых частиц. Редкие доминирующие миссенс-варианты APOE также способствуют развитию дисбеталипопротеинемии. Khalil YA, et al. (2021) сообщают о том, что у нескольких пациентов с СКГЛ выявились редкие патогенные варианты APOE [42]. Вопрос о том, могли ли это быть случаи дисбеталипопротеинемии, а не СКГЛ, остается открытым. Вариант Leu167del гена APOE был обнаружен у нескольких пациентов с СКГЛ и был связан с изолированной гиперхолестеринемией у их родственников [43]. Независимое когортное исследование показало наличие APOE варианта Leu167del у нескольких пациентов с аутосомно-доминантной гиперхолестеринемией [16], что делает ее связь с СКГЛ еще более неопределенной [44].
Almeda-Valdes P, et al. (2014) установили, что дополнительная площадь под кривой постпрандиальной липемии у пациентов с СКГЛ определяется уровнями апоВ-48 натощак и усиливается наличием абдоминального ожирения. Это исследование также послужило основанием для предположения, что ген APOA5, наряду с кластером APOA1/APOA4, связан с продукцией ХС-ЛОНП и хиломикронов у пациентов с СКГЛ [45]. Результаты исследования Di Taranto MD, et al. (2015) показали, что ОНВ S19W в гене APOA5 ассоциирован с СКГЛ независимо от уровня ОХС, ТГ и индекса массы тела [46].
В недавнем исследовании Taghizadeh E, et al. (2020) показан вариант, при котором аспарагиновая кислота заменяется аспарагином в позиции 151 в гене LPL (D151N), у пациентов с наличием СКГЛ. У пациентов с СКГЛ отмечается замедленный клиренс хиломикронов и остатков ХС-ЛОНП и одним из генов, участвующим в путях их выведения, является ген LPL [21].
Варианты потери функции гена ANGPTL3 являются причиной для моногенного заболевания — семейной комбинированной гиполипидемии, противоположному СКГЛ фенотипу. Исследователи предположили, что варианты усиления функции при ANGPTL3 могут привести к неблагоприятному повышению уровня липидов, т. е. повышению уровня ТГ и ХС-ЛНП. Однако недавнее исследование, проведенное Bea AM, et al. (2021), в результате которого был проведен скрининг кодирующих областей гена ANGPTL3 у 162 неродственных пациентов с СКГЛ, не выявило вариантов усиления функции [47].
Ученые со всего мира исследуют и предлагают множество других отдельных генов, которые могут играть роль в развитии СКГЛ, такие как LCAT, PPARA, TNFRSF1B, GPR77, PPARG, RXRG, LIPC, ATF6, PCSK9 [3][5][20][24][25][29][31][34][48][49]. Многие из них косвенно вовлечены в метаболические процессы, такие как нарушения функции жировой ткани, дефектный клиренс богатых ТГ липопротеинов и частиц ЛНП, увеличение синтеза ХС-ЛОНП и жира в печени и аномальный транспорт митохондриальных мембран [20]. Luo X, et al. (2015), обследовав 12 пациентов с СКГЛ, обнаружили 879 генов, включая 394 гена с повышенной регуляцией и 485 генов с пониженной регуляцией, которые при биоинформационном анализе фокусируются вокруг различных путей, имеющих отношение к ДЛП и атеросклерозу [50]. Ученым предстоит большой объем работы по определению вклада редких вариантов в этих многочисленных генах в патогенез СКГЛ. Всестороннее исследование моногенных и полигенных факторов в больших когортах пациентов с СКГЛ, возможно, послужит следующим этапом в понимании молекулярно-генетических механизмов при СКГЛ.
Несмотря на высокую распространенность и потенциальные негативные последствия для здоровья, СКГЛ часто не диагностируется, и пациенты не получают адекватной липидснижающей терапии.
Крайне важно выявлять лиц с СКГЛ из группы пациентов с ДЛП и как можно раньше проводить профилактику ССЗ из-за наличия комплекса рисков атеросклероза, связанных с наличием обилия мелких плотных частиц ЛНП, повышенного уровня апоВ, пониженного уровня ХС-ЛВП, хронического воспаления, резистентности к инсулину и сниженного клиренса остатков липопротеидов, богатых ТГ.
Актуальными являются также исследования, направленные на изучение лежащих в основе СКГЛ генетических и метаболических механизмов и на разработку эффективных стратегий лечения.
Отношения и деятельность. Работа выполнена в рамках темы Государственного задания № FWNR-
2022-0003.
1. Ежов М. В., Кухарчук В. В., Сергиенко И. В. и др. Нарушения липидного обмена. Клинические рекомендации 2023. Российский кардиологический журнал. 2023;28(5):5471. doi:10.15829/1560-4071-2023-5471.
2. Ballantyne C. M. Clinical Lipidology. 2023. Third Edition. ISBN: 978-0-323-88286-6. doi:10.1016/C2019-0-03574-1.
3. Trinder M, Vikulova D, Pimstone S, et al. Polygenic architecture and cardiovascular risk of familial combined hyperlipidemia. Atherosclerosis. 2022;340:35-43. doi:10.1016/j.atherosclerosis.2021.11.032.
4. Голубева О. А., Творогова М. Г., Малышев П. П. и др. Диагностика и лечение семейной комбинированной гиперлипидемии. Атеросклероз и дислипидемии. 2011;(3):52-5.
5. Gill PK, Hegele RA. Familial combined hyperlipidemia is a polygenic trait. Curr Opin Lipidol. 2022;33(2):126-32. doi:10.1097/MOL.0000000000000796.
6. Padda IS, Fabian D, Johal GS. Familial Combined Hyperlipidemia. 2023 Jun 3. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan.
7. Goldstein JL, Schrott HG, Hazzard WR, et al. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52(7):1544-68. doi:10.1172/JCI107332.
8. Bello-Chavolla OY, Kuri-García A, Ríos-Ríos M, et al. Familial combined hyperlipidemia: current knowledge, perspectives, and controversies. Rev Invest Clin. 2018;70(5):224-36. doi:10.24875/RIC.18002575.
9. Veerkamper MJ, de Graaf J, Bredie SJ, et al. Diagnosis of familial combined hyperlipidemia based on lipid phenotype expression in 32 families: results of a 5-year follow-up study. Arterioscler Thromb Vasc Biol. 2002;22(2):274-82. doi:10.1161/hq0202.104059.
10. Vaverková H, Karásek D. Familial combined hyperlipidemia — the most common genetic dyslipidemia in population and in patients with premature atherothrombotic cardiovascular disease. Vnitr Lek. 2018;64(1):25-9.
11. Brouwers MCGJ, de Graaf J, Simons N, et al. Incidence of type 2 diabetes in familial combined hyperlipidemia. BMJ Open Diabetes Res Care. 2020;8(1):e001107. doi:10.1136/bmjdrc-2019-001107.
12. Mandraffino G, Morace C, Franzè MS, et al. Fatty Liver as Potential Biomarker of Atherosclerotic Damage in Familial Combined Hyperlipidemia. Biomedicines. 2022;10(8):1770. doi:10.3390/biomedicines10081770.
13. Díaz-Ruiz M, Martínez-Triguero ML, López-Ruiz A, et al. Metabolic disorders and inflammation are associated with familial combined hyperlipemia. Clin Chim Acta. 2019;490:194-9. doi:10.1016/j.cca.2018.09.009.
14. Skoumas I, Andrikou I, Grigoriou K, et al. Lipoprotein(a), metabolic profile and new-onset type 2 diabetes in patients with familial combined hyperlipidemia: A 9 year follow-up study. J Clin Lipidol. 2023;17(4):512-8. doi:10.1016/j.jacl.2023.05.103.
15. Vargas-Vázquez A, Bello-Chavolla OY, Antonio-Villa NE, et al. Comparative assessment of LDL-C and VLDL-C estimation in familial combined hyperlipidemia using Sampson's, Martin's and Friedewald's equations. Lipids Health Dis. 2021;20(1):46. doi:10.1186/s12944-021-01471-3.
16. Martin SS. Calculating LDL cholesterol in familial combined hyperlipidemia: Out with the old, in with the new? Atherosclerosis. 2018;277:172-4. doi:10.1016/j.atherosclerosis.2018.07.034.
17. Zubirán R, Vargas-Vazquez A, Olvera FDR, et al. Performance of the enhanced Sampson-NIH equation for VLDL-C and LDL-C in a population with familial combined hyperlipidemia. Atherosclerosis. 2023;386:117364. doi:10.1016/j.atherosclerosis.2023.
18. Vikulova DN, Trinder M, Mancini GBJ, et al. Familial Hypercholesterolemia, Familial Combined Hyperlipidemia, and Elevated Lipoprotein(a) in Patients With Premature Coronary Artery Disease. Can J Cardiol. 2021;37(11):1733-42. doi:10.1016/j.cjca.2021.08.012.
19. Rallidis LS, Kosmas N, Tsirebolos G, et al. Prevalence of heterozygous familial hypercholesterolemia and combined hyperlipidemia phenotype in very young survivors of myocardial infarction and their association with the severity of atheromatous burden. J Clin Lipidol. 2019;13(3):502-8. doi:10.1016/j.jacl.2019.02.007.
20. Taghizadeh E, Esfehani RJ, Sahebkar A, et al. Familial combined hyperlipidemia: An overview of the underlying molecular mechanisms and therapeutic strategies. IUBMB Life. 2019;71(9):1221-9. doi:10.1002/iub.2073.
21. Taghizadeh E, Ghayour-Mobarhan M, Ferns GA, et al. A novel variant in LPL gene is associated with familial combined hyperlipidemia. Biofactors. 2020;46(1):94-9. doi:10.1002/biof.1570.
22. Luijten J, van Greevenbroek MMJ, Schaper NC, et al. Incidence of cardiovascular disease in familial combined hyperlipidemia: A 15-year follow-up study. Atherosclerosis. 2019;280:1-6. doi:10.1016/j.atherosclerosis.2018.11.013.
23. Kwiterovich PO, Coresh J, Smith HH, et al. Comparison of the plasma levels of apolipoproteins B and A-1, and other risk factors in men and women with premature coronary artery disease. Am J Cardiol. 1992;69:1015-1021.
24. Brahm AJ, Hegele RA. Combined hyperlipidemia: familial but not (usually) monogenic. Curr Opin Lipidol. 2016;27:131-40. doi:10.1097/MOL.0000000000000270.
25. Taghizadeh E, Farahani N, Mardani R, et al. Genetics of Familial Combined Hyperlipidemia (FCHL) Disorder: An Update. Biochem Genet. 2022;60(2):453-81. doi:10.1007/s10528-021-10130-2.
26. Pedro-Botet J, Climent E, Gabarró N, et al. Familial combined hyperlipidaemia/polygenic mixed hyperlipidaemia. Clin Investig Arterioscler. 2021;33 Suppl 2:43-49. English, Spanish. doi:10.1016/j.arteri.2020.12.013.
27. Dron JS, Hegele RA. Genetics of hypertriglyceridemia. Front Endocrinol. 2020;11:455. doi:10.3389/fendo.2020.00455.
28. Ripatti P, Rämö JT, Mars NJ, et al. Polygenic hyperlipidemias and coronary artery disease risk. Circ Genom Precis Med. 2020;13:e002725. doi:10.1161/CIRCGEN.119.002725.
29. Ripatti P, Rämö JT, Söderlund S, et al. The contribution of GWAS loci in familial dyslipidemias. PLoS Genet. 2016;12:e1006078. doi:10.1371/journal.pgen.1006078.
30. Gill PK, Dron JS, Berberich AJ, et al. Combined hyperlipidemia is genetically similar to isolated hypertriglyceridemia. J Clin Lipidol. 2021;15:79-87. doi:10.1016/j.jacl.2020.11.006.
31. Dron JS, Wang J, McIntyre AD, et al. The polygenic nature of mild-to-moderate hypertriglyceridemia. J Clin Lipidol. 2020;14(1):28-34.e2. doi:10.1016/j.jacl.2020.01.003.
32. Berberich AJ, Hegele RA. A Modern Approach to Dyslipidemia. Endocr Rev. 2022;43(4):611-53. doi:10.1210/endrev/bnab037.
33. Baila-Rueda L, Cenarro A, Lamiquiz-Moneo I, et al. Cholesterol oversynthesis markers define familial combined hyperlipidemia versus other genetic hypercholesterolemias independently of body weight. J Nutr Biochem. 2018;53:48-57. doi:10.1016/j.jnutbio.2017.10.005.
34. Borén J, Packard CJ, Taskinen MR. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front Endocrinol (Lausanne). 2020;11:474. doi:10.3389/fendo.2020.00474.
35. Taghizadeh E, Mirzaei F, Jalilian N, et al. A novel mutation in USF1 gene is associated with familial combined hyperlipidemia. IUBMB Life. 2020;72(4):616-23. doi:10.1002/iub.2186.
36. Pajukanta P, Lilja HE, Sinsheimer JS, et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1). Nat Genet. 2004;36(4):371-6. doi:10.1038/ng1320.
37. Laurila PP, Soronen J, Kooijman S, et al. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016;8(323):323ra13. doi:10.1126/scitranslmed.aad0015.
38. Taghizadeh E, Mardani R, Rostami D, et al. Molecular mechanisms, prevalence, and molecular methods for familial combined hyperlipidemia disease: A review. J Cell Biochem. 2019;120(6):8891-8. doi:10.1002/jcb.28311.
39. Naukkarinen J, Nilsson E, Koistinen HA, et al. Functional variant disrupts insulin induction of USF1: mechanism for USF1-associated dyslipidemias. Circ Cardiovasc Genet. 2009;2(5):522-9. doi:10.1161/CIRCGENETICS.108.840421.
40. Auer S, Hahne P, Soyal SM, et al. Potential role of upstream stimulatory factor 1 gene variant in familial combined hyperlipidemia and related disorders. Arterioscler Thromb Vasc Biol. 2012;32(6):1535-44. doi:10.1161/ATVBAHA.112.245639.
41. Bea AM, Larrea-Sebal A, Marco-Benedi V, et al. Contribution of APOE Genetic Variants to Dyslipidemia. Arterioscler Thromb Vasc Biol. 2023;43(6):1066-77. doi:10.1161/ATVBAHA.123.318977.
42. Khalil YA, Rabès JP, Boileau C, et al. APOE gene variants in primary dyslipidemia. Atherosclerosis. 2021;328:11-22. doi:10.1016/j.atherosclerosis.2021.05.007.
43. Marduel M, Ouguerram K, Serre V, et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum Mutat. 2013;34(1):83-7. doi:10.1002/humu.22215.
44. Solanas-Barca M, de Castro-Orós I, Mateo-Gallego R, et al. Apolipoprotein E gene mutations in subjects with mixed hyperlipidemia and a clinical diagnosis of familial combined hyperlipidemia. Atherosclerosis. 2012;222(2):449-55. doi:10.1016/j.atherosclerosis.2012.03.011.
45. Almeda-Valdes P, Cuevas-Ramos D, Mehta R, et al. Factors associated with postprandial lipemia and apolipoprotein A-V levels in individuals with familial combined hyperlipidemia. BMC Endocr Disord. 2014;14:90. doi:10.1186/1472-6823-14-90.
46. Di Taranto MD, Staiano A, D'Agostino MN, et al. Association of USF1 and APOA5 polymorphisms with familial combined hyperlipidemia in an Italian population. Mol Cell Probes. 2015;29(1):19-24. doi:10.1016/j.mcp.2014.10.002.
47. Bea AM, Franco-Marín E, Marco-Benedí V, et al. ANGPTL3 gene variants in subjects with familial combined hyperlipidemia. Sci Rep. 2021;11(1):7002. doi:10.1038/s41598-021-86384-y.
48. Luo X, Yu C, Fu C, et al. Identification of the differentially expressed genes associated with familial combined hyperlipidemia using bioinformatics analysis. Mol Med Rep. 2015;11(6):4032-8. doi:10.3892/mmr.2015.3263.
49. Li Z, Zhang X, Li X, et al. A nonintegrated iPSC line (SDQLCHi042-A) from a boy suffering from familial combined hyperlipidemia with compound heterozygous mutations of lipoprotein lipase gene. Stem Cell Res. 2021;53:102313. doi:10.1016/j.scr.2021.102313.
50. Shakhtshneider E, Ivanoshchuk D, Timoshchenko O, et al. Analysis of Rare Variants in Genes Related to Lipid Metabolism in Patients with Familial Hypercholesterolemia in Western Siberia (Russia). J Pers Med. 2021;11:1232. doi:10.3390/jpm11111232.
Тимощенко Ольга Владимировна — к. м. н., н. с. лаборатории молекулярно-генетических исследований терапевтических заболеваний, врач-кардиолог.
Новосибирск
Нет
Шахтшнейдер Елена Владимировна — к. м. н., руководитель сектора изучения моногенных форм распространенных заболеваний человека, ИЦиГ СО РАН; зам. руководителя филиала по научной работе, НИИТПМ – филиал ИЦиГ СО РАН.
Новосибирск
Нет
Тимощенко О.В., Шахтшнейдер Е.В. Семейная комбинированная гиперлипидемия, современное состояние проблемы (обзор литературы). Российский кардиологический журнал. 2024;29(8):5874. https://doi.org/10.15829/1560-4071-2024-5874. EDN: FQIHBM
Timoshchenko O.V., Shakhtshneider E.V. Familial combined hyperlipidemia: current status of the problem (literature review). Russian Journal of Cardiology. 2024;29(8):5874. (In Russ.) https://doi.org/10.15829/1560-4071-2024-5874. EDN: FQIHBM













































