



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2021-4209
Перейти к:
Легочная гипертензия представляет собой сложный для дифференциальной диагностики синдром, являющийся исходом различных патологических состояний. При исключении наиболее распространенных причин развития легочной гипертензии — патологии левых камер сердца и тромбоэмболии легочной артерии — дальнейший поиск этиологии зачастую становится неразрешимой проблемой. Синдром Сезари относят к редким клиническим вариантам Т-клеточных злокачественных лимфом кожи. Важное значение имеет ранняя диагностика этого синдрома для начала адекватной терапии, т.к. случаи полной реконвалесценции или длительной ремиссии у больных синдромом Сезари казуистически редки. Описан клинический случай верификации синдрома Сезари в практике кардиологического отделения.
Кижватова Н.В., Космачева Е.Д., Кручинова С.В., Намитоков А.М. Случай сложной диагностики редкой причины легочной гипертензии. Российский кардиологический журнал. 2021;26(1S):4209. https://doi.org/10.15829/1560-4071-2021-4209
Kizhvatova N.V., Kosmacheva E.D., Kruchinova S.V., Namitokov A.M. Difficult diagnostics of a rare cause of pulmonary hypertension: a case report. Russian Journal of Cardiology. 2021;26(1S):4209. (In Russ.) https://doi.org/10.15829/1560-4071-2021-4209
Интерес к изучению причин формирования и подходов к лечению легочной гипертензии (ЛГ) существенно возрос в последние 2 десятилетия. Это связано с расширением как диагностических методик, так и терапевтических стратегий лечения, что способствовало существенному прогрессу в улучшении качества и длительности жизни пациентов с ЛГ. В классификации причин ЛГ Европейского общества кардиологов (2015) выделяют 5 основных групп [1]:
1) Легочная артериальная гипертензия (ЛАГ);
2) ЛГ, обусловленная поражением левых камер сердца;
3) ЛГ, обусловленная патологией легких и/или гипоксией;
4) ЛГ из-за обструкции легочной артерии (ЛА);
5) ЛГ, обусловленная неясными многофакторными механизмами.
Последняя (пятая) группа представляет собой достаточно разнородный по своим этиологическим и патогенетическим проявлениям набор заболеваний, редко встречающихся в практике врача-кардиолога.
В представленной работе разбирается поэтапный диагностический анализ крайне редкого случая развития ЛГ при миелопролиферативном заболевании.
Описание клинического случая
Пациентка В., 50 лет, доставлена в клинику с жалобами на выраженную слабость, одышку при минимальной физической нагрузке и в покое, сердцебиение, перебои в работе сердца, повышение артериального давления до 160 170/95 мм рт.ст.; увеличение живота в объеме; боли в суставах; выраженную сухость и шелушение кожи.
Из анамнеза известно, что описанные жалобы имелись в течение одного года с прогрессирующим снижением толерантности к физическим нагрузкам. При сборе анамнеза жизни установлено, что 2 года назад пациентка обследовалась у гинеколога по поводу миомы матки, были выявлены анемия, лейкоцитоз, тромбоцитоз. В течение 2-х лет похудела на 18 кг. Около 1 года беспокоят боли в разных суставах, выраженная слабость, увеличение периферических лимфатических узлов различной локализации (шейных, затылочных, паховых, подмышечных). Консультирована ревматологом и гематологом: диагностирован деформирующий артроз стоп, высказано предположение о лимфопролиферативном заболевании, в связи с чем была выполнена стернальная пункция и биопсия лимфатического узла, по данным которых признаков острого и хронического гемобластоза не получено. Назначена терапия витамином В12 и фолиевой кислотой, на фоне которой анемия скорректирована.
Данные объективного осмотра. Общее состояние: тяжелое. Астеническое телосложение, истощена. Рост — 160 см, вес — 40 кг. Индекс массы тела — 15,62 кг/м2. Кожные покровы: сухие; гиперпигментация кожи, мелко- и крупнопластическое шелушение; умеренный акроцианоз (рис. 1). Увеличены подчелюстные и подмышечные лимфоузлы. Видимые слизистые оболочки: бледные, цианотичные. Подкожная клетчатка практически отсутствует. Отечность голеней и стоп; отечность передней брюшной стенки, асцит. Сердечно-сосудистая система: ритм правильный с частотой сердечных сокращений (ЧСС) 98 уд./мин. 2 тон — акцентирован на ЛА, определяется мягкий систолический шум на верхушке; пульс 98 уд./мин, ритмичный, слабого наполнения, артериальное давление: 100/60 мм рт.ст. Органы дыхания: грудная клетка — правильной формы; дыхание везикулярное, ослабленное; хрипы не выслушиваются; частота дыхания 20 в 1 мин. Органы пищеварения: язык — сухой, обложен белым налетом. Живот увеличен, при пальпации болезненный в правом подреберье, определяется свободная жидкость в брюшной полости. Печень: пальпаторно увеличена — нижний край на 2,5-3 см выступает из-под реберной дуги; край плотный, ровный, размер по Курлову 12×10×9 см. Опорно-двигательный аппарат: деформация суставов (рис. 2). Мочеполовая система: Симптом поколачивания отрицательный с обеих сторон.
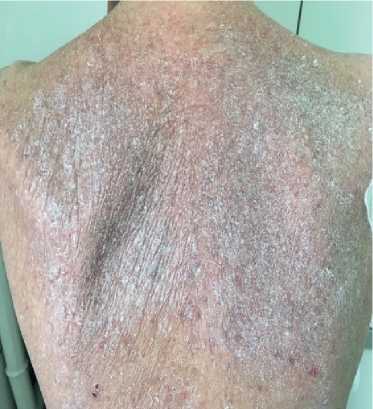
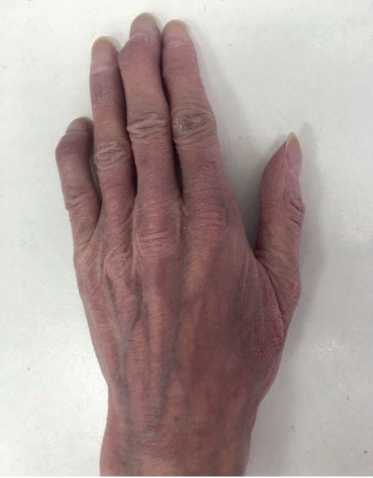
Таким образом, у пациентки имели место следующие симптомокомплексы: 1) отечный (асцит, отеки нижних конечностей); 2) гепатомегалия; 3) кожный; 4) суставной; 5) дыхательная недостаточность; 6) лимфаденопатия.
По данным инструментальных методов исследования выявлены следующие изменения:
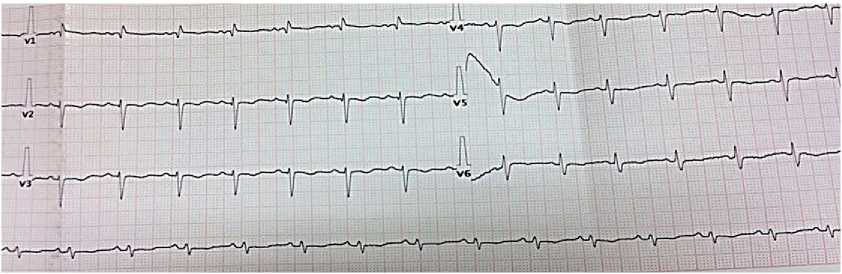
Электрокардиограмма — синусовая тахикардия с ЧСС 100 уд./мин. Электрическая ось сердца отклонена вправо. Снижен вольтаж зубцов. Нарушение проводимости по правой ножке пучка Гиса. Признаки нагрузки на правый желудочек (ПЖ) (рис. 3).
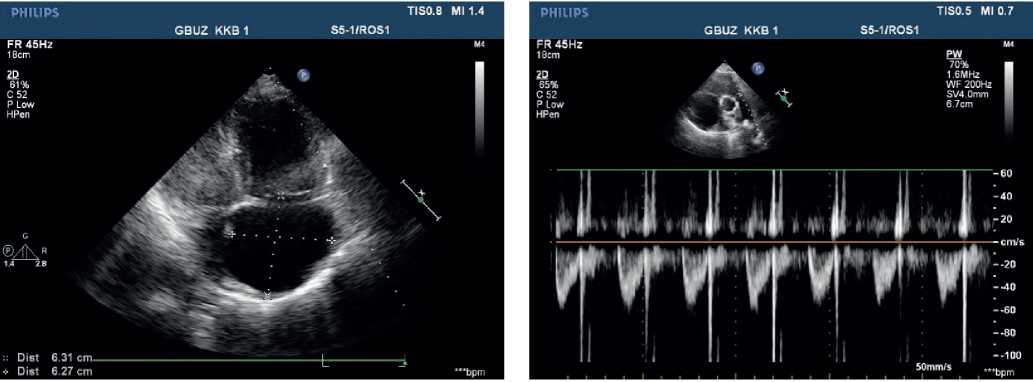
Эхокардиография (ЭхоКГ) — аорта: восходящий отдел: 30 мм Регургитация на аортальном клапане: ++. Левое предсердие 29 мм. Конечный диастолический размер левого желудочка (ЛЖ): 38 мм, межжелудочковая перегородка (МЖП) 9 мм, задняя стенка (ЗС) ЛЖ 10 мм. Фракция выброса ЛЖ (по Симпсону): 43%. Диффузный гипокинез стенок ЛЖ с парадоксальным движением МЖП. Допплерография митрального клапана: +. Без выраженной реакции на фазы дыхания. Правое предсердие (ПП) 55×58 мм. ПЖ 32 мм. Приточный отдел ПЖ 53 мм. Диффузный гипокинез стенок ПЖ. Верхушка сердца выполнена преимущественно ПЖ. Трикуспидальный клапан (ТК) створки: уплотнены утолщены. Прогиб створок ТК в полость ПП. Допплерография ТК: +++, (+++/++++) ЛА: с признаками выраженной гипертензии. Давление диастолическое: 25 мм рт.ст. Систолическое давление 90 мм рт.ст. Допплерография ЛА: ++. Нижняя полая вена: 20 мм, реакция неадекватная, с эффектом спонтанного контрастирования. По всему периметру сердца эхо-свободное пространство: перед ПЖ из эпигастрального доступа 12 мм, над ПП 5 мм, у верхушки ЛЖ 5 мм, за боковой стенкой 12 мм, за ЗС ЛЖ 17 мм (рис. 4, 5).
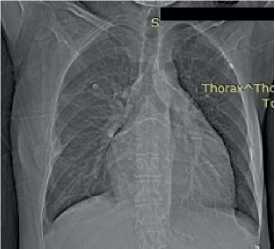
Рентгенография органов грудной клетки — легкие без очаговых и инфильтративных изменений. Кардиомегалия 3 ст., кардиоторакальный индекс 52% (рис. 6).
На основании вышеописанных данных дифференциально-диагностический ряд включал: онкологическое заболевание с паранеопластическим синдромом, патологию сердечно-сосудистой системы, патологию легких с формированием легочного сердца и хронической сердечной недостаточности. Не исключалось сочетание нескольких заболеваний.
Пациентке выполнены фиброгастроскопия, колоноскопия, ультразвуковое исследование гениталий, данных за новообразование не выявлено.
При анализе ЭхоКГ обратило внимание высокое значение давления в ЛА и расширение правых отделов сердца в отсутствие структурной патологии левых камер сердца.
Согласно клиническим рекомендациям по диагностике и лечению пациентов с ЛГ, наиболее часто встречается ЛГ вследствие патологии левых камер сердца [1].
Для исключения патологии левых отделов сердца как возможной причины ЛГ было выполнено зондирование правых отделов сердца катетером СванГанса, по данным которого давление заклинивания в ЛА — 13 мм рт.ст., систолическое давление в ЛА — 48 мм рт.ст., легочное сосудистое сопротивление — 380 мм; индекс легочного сосудистого сопротивления — 412, что говорит в пользу прекапиллярной ЛГ.
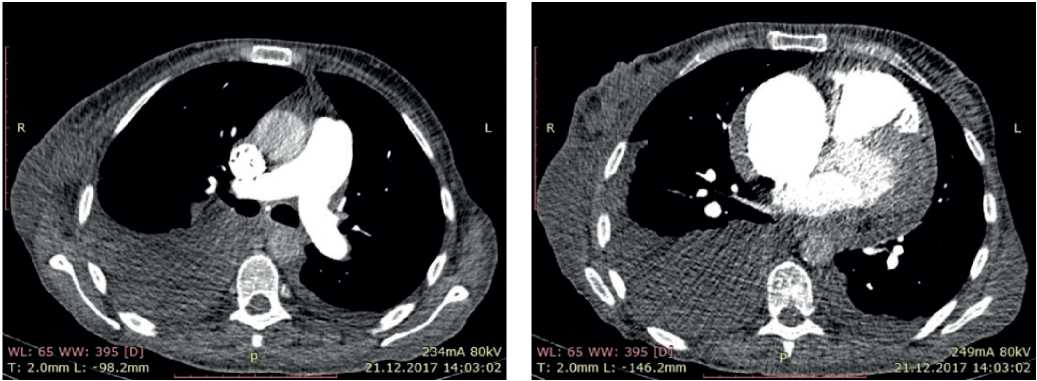
С целью исключения легочной патологии выполнена компьютерная томография (КТ) легких и оценена функция внешнего дыхания (ФВД). КТ органов грудной клетки: структурной патологии легких не выявлено. ФВД: умеренные нарушения по рестриктивному типу. Учитывая возможность наличия высокой ЛГ на фоне хронической тромбоэмболической болезни легких, выполнена КТ ЛА с контрастированием, по данным которой признаков тромбоза не выявлено (рис. 7).
Как известно, ЛАГ наиболее часто может быть обусловлена наличием врожденных пороков сердца (дефекты межпредсердной перегородки/МЖП, открытый артериальный проток, частичный аномальный дренаж легочных вен), а также ВИЧ-инфекцией и портальной гипертензией. По данным чреспищеводной ЭхоКГ, КТ сердца с контрастированием, ультразвукового исследования органов брюшной полости и лабораторных исследований данные причины были исключены, наличие ВИЧ-инфекции также не выявлено.
Возраст пациентки на момент развития симптомов и наличие сопутствующих синдромов системного поражения (кожный, кахексия, лимфаденопатия) делало маловероятным наследственный характер ЛГ.
В последующем диагностический поиск был направлен на выявление патологий, входящих в группу ЛГ с неясными и/или множественными механизмами. Наличие лимфаденопатии, суставного синдрома, гепатомегалии позволило заподозрить наличие лимфопролиферативного заболевания.
Данные морфологического и иммунногистохимического анализа биоптатов:
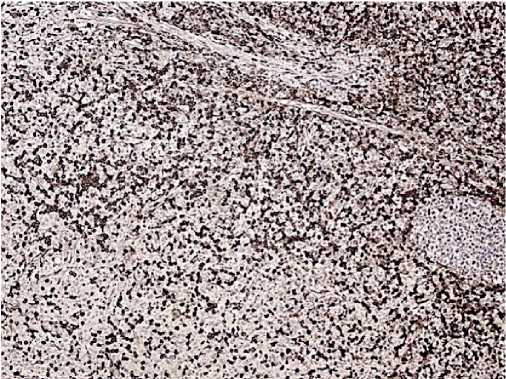
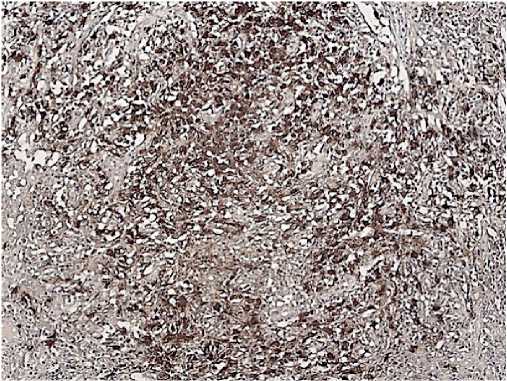
Морфологически: кожа — эпидермис с гиперкератозом, неравномерно истончен, в базальном слое лимфоидный инфильтрат с педжетоидным типом роста, выраженным экзоцитозом, представлен клетками среднего размера с ядрами неправильной формы, выраженной складчатостью ядерной мембраны. Лимфоузлы — структура лимфоузлов нарушена за счет резкого расширения паракортикальной зоны, инфильтрированной полигональными клетками
с ядрами неправильной формы, складчатой ядерной мембраной. Предсуществующие фолликулы мелкие с истонченной мантийной зоны, оттеснены опухолевым инфильтратом. Синусы расширены. Мозговой слой с липоматозом. (серия биопсий представлена на рисунках 8-10). Заключение гистологического исследования: иммуноморфологическая картина соответствует синдрому Сезари с поражением лимфатических узлов. Диагноз: Синдром Сезари — Т клеточная лимфома с поражением лимфатических узлов.
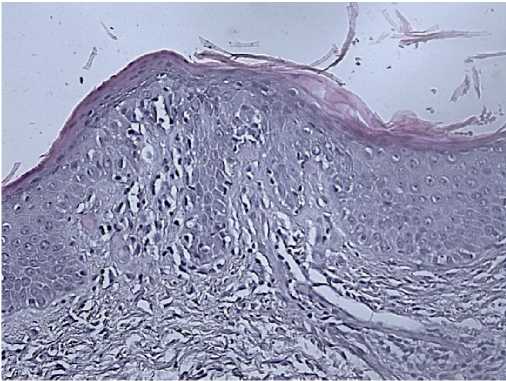
Заключительный диагноз: Синдром Сезари (grade III), II клин. группа. Высокая ЛАГ на фоне лимфопролиферативного заболевания (синдром Сезари). Хроническое легочное сердце, хроническая сердечная недостаточность II Б стадии.
На фоне проводимой терапии (диуретическая, пульсурежающая) увеличилась толерантность к физической нагрузке, регрессировал отечный синдром. Спустя 4 года динамического наблюдения пациентка жива, чувствует себя удовлетворительно, получает рекомендуемую терапию.
Синдром Сезари — крайне редкое заболевание — 3 случая на 1 млн населения [2][3], которое является вариантом Т-клеточной лимфомы (вовлечены CD4+ Т-лимфоциты периферические) и характеризуется генерализованной эритродермией, лимфаденопатией и наличием клеток Сезари в коже, лимфатических узлах и периферической крови [3][4].
Важное значение имеет ранняя диагностика этого синдрома для начала адекватной терапии, т.к. случаи полной реконвалесценции или длительной ремиссии у больных синдромом Сезари казуистически редки [5].
Согласно литературным данным, случаи высокой ЛГ описываются у пациентов с В-клеточной лимфомой. Подобные случаи при Т-клеточных лимфомах крайне редки [4][5].
Представленный клинический случай, согласно последней классификации, относится к пятой группе причин ЛГ.
По классификации лимфоидных опухолей ВОЗ (2008) грибовидный микоз/синдром Сезари относится к первичным Т-клеточным лимфомам кожи из зрелых (периферических) клеток [2]. Заболевание впервые описал французский дерматолог Алибер в 1806г. Он же в 1832г предложил название, подчеркнув характерные клинические проявления болезни, — грибовидные опухоли. В 1879г французский дерматолог Базен подробно описал клинику классической формы заболевания, и с тех пор оно носит имя Алибера-Базена. Первое описание болезни в России сделал А.М. Стуковенков в 1889г [1][5]. Французский дерматолог Сезари в 1938г впервые описал 59-летнюю женщину с генерализованной эритродермией, лимфаденопатией, интенсивным зудом и наличием в периферической крови “чудовищных”, “уродливых” моноцитоидных клеток [1]. Синдром Сезари составляет 2-3% всех злокачественных лимфом [2]. Он рассматривается как лейкемический вариант заболевания, характеризующийся хронической эритродермией с диффузной лимфоидной инфильтрацией дермы, выходом аномальных лимфоцитов в кровь и поражением костного мозга [4]. Классические клетки Сезари имеют крупные складчатые ядра, сходные с головным мозгом, в связи с чем и получили название “церебриформных” или “мозговидных” клеток. В основном они локализуются в верхних слоях дермы и эпидермисе и имеют иммунологический фенотип Т-хелперов (СD2+, CD3+, CD4+, CD5+) [2].
Данный диагноз устанавливается на основании комплексной оценки клинической картины заболевания, гистологического и иммунофенотипического исследования биоптатов из очагов поражения кожи, определения перестройки гена Т-клеточного рецептора. Необходимо отметить, что в настоящее время не существует единых общепринятых диагностических критериев грибовидного микоза, а клинические руководства значительно различаются между собой в объеме рекомендуемых исследований, необходимых для постановки диагноза. Клиническое обследование пациента остается основополагающим методом в диагностике, т.к. дает возможность определить разновидность и стадию этого заболевания, но при лейкемической форме Т-клеточной лимфомы кожи возрастает значимость обычного клинического анализа периферической крови [5].
Поражение легких при Т-клеточных лимфомах встречается примерно в 10% случаев [6]. Основным механизмом развития ЛГ у таких пациентов считается стимуляция эндотелиальных клеток сосудов факторами, связанными с тромбоцитами — в первую очередь серотонином и фактором роста [7]. Другими вероятными причинами рассматриваются повышенная вязкость крови при аномалиях функции иммуноглобулинов [8][9] и микроэмболия опухолевыми клетками [8].
В представленном клиническом случае продемонстрирован последовательный алгоритм диагностических шагов, позволивший установить редкую причину ЛГ. Учитывая нечастое упоминание синдрома Сезари в кардиологической литературе, объединение разобщенных клинических синдромов, характерных для данного заболевания, в единую нозологию представляет диагностическую сложность.
Отсутствие рекомендаций по терапии ЛГ у пациентов с лимфопролиферативными заболеваниями требует индивидуального подхода к назначению ЛАГ-специфической терапии в каждом конкретном случае.
1. Galie N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016:37(1):67-119. doi:10.1093/eurheartj/ehv317.
2. Чазова И. Е., Мартынюк Т. В. Легочная гипертензия. М.:Практика. 2015:450-67. ISBN: 978-5-89816-138-5
3. LeRoy EC, Black C, Fleischmajer R, et al. Scl eroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202-5.
4. Mukerjee D, St George D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2013;62(11):1088-93. doi:10.1136/ard.62.11.1088.
5. MacGregor AJ, Canavan R, Knight C, et al. Pulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survival. Rheumatology (Oxford). 2001;40(4):453-9. doi:10.1093/rheumatology/40.4.453.
6. de Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol. 2010;148(5):673-89. doi:10.1111/j.1365-2141.2009.08003.x.
7. Snyder LS, Harmon KR, Estensen RD. Intravascular lymphomatosis (malignant angio-endotheliomatosis) presenting as pulmonary hypertension. Chest. 1989;96(5):1199-200. doi:10.1378/chest.96.5.1199.
8. Aouba A, Diop S, Saadoun D, et al. Severe pulmonary arterial hypertension as initial manifestation of intravascular lymphoma: case report. Am J Hematol. 2005;79(1):46-9. doi:10.1002/ajh.20300.
9. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655-65. doi:10.1056/NEJMra035488.
Заведующая кардиологическим отделением № 3; доцент кафедры терапии № 1 ФПК и ППС.
Краснодар.
Нет
Доктор медицинских наук, заместитель главного врача по лечебной работе; заведующая кафедрой терапии № 1 ФПК и ППС.
350086, Краснодар, ул. 1 Мая, 167.
Нет
Врач кардиологического отделения № 1; аспирант кафедры терапии № 1 ФПК и ППС.
Краснодар.
Нет
Заведующий кардиологическим отделением № 2; ассистент кафедры терапии № 1 ФПК и ППС.
Краснодар.
Нет
Кижватова Н.В., Космачева Е.Д., Кручинова С.В., Намитоков А.М. Случай сложной диагностики редкой причины легочной гипертензии. Российский кардиологический журнал. 2021;26(1S):4209. https://doi.org/10.15829/1560-4071-2021-4209
Kizhvatova N.V., Kosmacheva E.D., Kruchinova S.V., Namitokov A.M. Difficult diagnostics of a rare cause of pulmonary hypertension: a case report. Russian Journal of Cardiology. 2021;26(1S):4209. (In Russ.) https://doi.org/10.15829/1560-4071-2021-4209













































