


Содержание
Перейти к:
 О. С. Чумакова,
Л. Э. Кертиева,
Л. О. Минушкина,
Л. В. Ухова,
Д. М. Алькам,
З. И. Алиева,
Н. А. Ильенкова,
Т. С. Архипкина,
Н. М. Волошина,
Т. Н. Бакланова,
Д. А. Затейщиков
О. С. Чумакова,
Л. Э. Кертиева,
Л. О. Минушкина,
Л. В. Ухова,
Д. М. Алькам,
З. И. Алиева,
Н. А. Ильенкова,
Т. С. Архипкина,
Н. М. Волошина,
Т. Н. Бакланова,
Д. А. Затейщиков https://doi.org/10.15829/1560-4071-2025-6412
EDN: ZEUPAG
Перейти к:
Цель. Оценить частоту встречаемости амилоидоза сердца и болезни Фабри среди взрослых пациентов с фенотипом ГКМП в российском наблюдательном исследовании и сравнить результаты с международными данными на основе мета-анализа.
Материал и методы. В проспективное исследование, проводимое в ГБУЗ ГКБ №17 ДЗМ с 2009 по 2024гг, включены 223 больных с фенотипом ГКМП (средний возраст 54±14,9 лет, 55% мужчин). Все пациенты прошли клинико-инструментальное, лабораторное и генетическое обследование. Диагнозы фенокопий верифицировались в соответствии с рекомендациями. Далее их долю в общей группе сравнивали с данными мета-анализа публикаций, отобранных в результате систематического поиска в базе PubMed по распространенности AL- и ATTRv-амилоидоза сердца и болезни Фабри среди взрослых пациентов с ГКМП.
Результаты. В нашей когорте диагноз был пересмотрен на AL-амилоидоз у шести больных (2,7%), на ATTRv-амилоидоз – у трех (1,4%) и на болезнь Фабри – у одного (0,5%). В мета-анализ включены 16 исследований (8243 пациента). Различий между российскими данными и данными других популяций не выявлено. Общая распространенность фенокопий по результатам мета-анализа с учетом российской когорты составила: AL-амилоидоз - 1,8%, ATTRv-амилоидоз - 1,2% и болезнь Фабри - 1,2%.
Заключение. Фенокопии составляют существенную долю причин фенотипа ГКМП у взрослых, а их распространенность в России сопоставима с другими популяциями. Повышенная клиническая настороженность и обязательное генетическое тестирование могут улучшить выявляемость редких заболеваний, маскирующихся под ГКМП.
Чумакова О.С., Кертиева Л.Э., Минушкина Л.О., Ухова Л.В., Алькам Д.М., Алиева З.И., Ильенкова Н.А., Архипкина Т.С., Волошина Н.М., Бакланова Т.Н., Затейщиков Д.А. Распространенность амилоидоза сердца и болезни Фабри среди взрослых больных с фенотипом гипертрофической кардиомиопатии: данные российского одноцентрового исследования и мета-анализ. Российский кардиологический журнал. 2025;30(10):6412. https://doi.org/10.15829/1560-4071-2025-6412. EDN: ZEUPAG
Chumakova O.S., Kertieva L.E., Minishkina L.O., Ukhova L.V., Alkam D.M., Alieva Z.I., Ilyenkova N.A., Arkhipkina T.S., Voloshina N.M., Baklanova T.N., Zateyshchikov D.A. Prevalence of cardiac amyloidosis and Fabry disease among adult patients with hypertrophic cardiomyopathy phenotype: data from a Russian single-center study and meta-analysis. Russian Journal of Cardiology. 2025;30(10):6412. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6412. EDN: ZEUPAG
Гипертрофическая кардиомиопатия (ГКМП) — преимущественно генетически обусловленное заболевание сердца, характеризующееся утолщением миокарда левого желудочка (ЛЖ) (у взрослых пробандов — ≥1,5 см), часто асимметричного характера с обструкцией выводного отдела ЛЖ, не объяснимое другими потенциально причинными заболеваниями. В половине случаев заболевание связывают с носительством патогенных вариантов (мутаций) в генах саркомерных белков [1].
Амилоидоз и болезнь Фабри — заболевания, которые могут имитировать ГКМП у взрослых, поэтому их часто называют "фенокопиями". Оценка их распространенности сильно варьирует в зависимости от характеристик пациентов и методов скрининга. В последнее десятилетие интерес к этим заболеваниям значительно возрос благодаря появлению специфической терапии. В российской популяции данные о частоте фенокопий среди взрослых с ГКМП ограничены и получены преимущественно в федеральных центрах, ориентированных на тяжелые случаи [2][3], что может завышать их распространенность. Исключением является исследование Savostyanov K, et al. (2022) [4], где скрининг на болезнь Фабри проводился в неспециализированных учреждениях. Повышение осведомленности врачей первичного звена о реальной частоте этих заболеваний в группах риска может улучшить их диагностику.
Целью работы было оценить частоту встречаемости амилоидоза сердца и болезни Фабри среди взрослых пациентов с фенотипом ГКМП в российском наблюдательном исследовании и сравнить результаты с международными данными на основе метаанализа.
В проспективное наблюдательное исследование, проводимое на базе ГБУЗ "Городская клиническая больница 17 ДЗМ" с 2009 по 2024гг, включены 223 неродственных больных (средний возраст 54±14,9 лет, 55% мужчин) с первичным диагнозом ГКМП, который устанавливали при эхокардиографии. Исключались больные с гипертрофией миокарда, обусловленной артериальной гипертензией, пороками сердца или спортивной адаптацией. Дальнейшее обследование проводилось в соответствии с ранее описанным протоколом [5] и включало регистрацию электрокардиограммы (ЭКГ) в покое и при суточном мониторировании по Холтеру, определение N-концевого промозгового натрийуретического пептида крови, магнитно-резонансную томографию сердца с оценкой отсроченного накопления гадолиний-содержащих препаратов. Панель генетического тестирования включала гены саркомера, транстиретина (transthyretin, TTR) и альфа-галактозидазы А (alpha galactosidase A, GLA). При наличии признаков альтернативной природы заболевания диагноз уточнялся в соответствии с рекомендациями [6] (Клинические рекомендации. Болезнь Фабри 2024).
Исследование одобрено этическим комитетом ГБУЗ "ГКБ № 17 ДЗМ" (протоколы № 5 от 22 мая 2009г и № 9 от 04 ноября 2016г). Все пациенты дали письменное согласие на участие.
Отбор публикаций для метаанализа
Отбор статей для метаанализа проведен независимо двумя исследователями в базе PubMed на английском языке по критериям PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analysis 2020). В случае разногласий решение принималось на основе консенсуса. Для каждой фенокопии использовались отдельные поисковые запросы. Включали работы, опубликованные между 01.01.2014 и 15.04.2025, в которых имелись данные о частоте как минимум одной из анализируемых фенокопий среди пациентов 18 лет и старше с гипертрофией миокарда ЛЖ ≥1,5 см. Все отобранные публикации проверялись вручную для оценки патогенности вариантов в генах TTR и GLA по критериям American College of Medical Genetics and Genomics (ACMG) и поиска релевантных публикаций в списках литературы. Исключались исследования, где диагноз фенокопии был установлен до включения, обзоры литературы и клинические случаи. В метаанализ каждой фенокопии включались данные нашего исследования.
Статистический анализ. Анализ данных проводили в SPSS 26.0 и MedCalc 23.1.3. Для количественных переменных применяли t-критерий Стьюдента при нормальном распределении или непараметрические методы при его отсутствии. Результаты представляли как среднее (M±SD) или медиану [ 25;75 процентили]. Дискретные переменные сравнивали с использованием критерия χ² Пирсона. Значимость устанавливали при p<0,05. Метаанализ выполняли по принципам MOOSE [7]. Для расчета суммарной доли использовали преобразование Фримана-Туки с моделями фиксированных и случайных эффектов. Результаты представляли на forest-plots. Гетерогенность оценивали по критериям Q и I². При высокой гетерогенности (I²>75%) применяли случайную модель, иначе — фиксированную. Гетерогенность по критерию Q считали значимой при p<0,1. Систематическую ошибку публикаций проверяли тестами Эггера (оценка смещения по размеру выборок) и Бегга (оценка дисперсии данных) с визуализацией на funnel-plots.
В исследование включены 223 неродственных больных с первичным диагнозом ГКМП, который был пересмотрен на амилоидоз легких цепей иммуноглобулинов (AL) у 6 (2,7%), на наследственный транстиретиновый амилоидоз (ATTRv) у 3 (1,4%) и на болезнь Фабри у 1 (0,5%). В 7 случаях амилоидоза (6 AL и 1 ATTRv) диагноз был заподозрен клинически, в остальных — на основании генетического тестирования.
Средний интервал между первоначальным диагнозом и верификацией составил 8 мес. (6 мес. для AL, 12 мес. для ATTRv). Двое больных с ATTRv были носителями TTR-варианта p.V50M, один — TTR-варианта p.E112K. Признаки застойной сердечной недостаточности отмечались у 6 (67%), рестриктивный тип диастолической дисфункции ЛЖ — у 4 (44%), гидроперикард — у 5 (56%). Обструкция выводного отдела ЛЖ отсутствовала у всех. Асимметричная форма ГКМП выявлена у 3 (33%) больных, низкий вольтаж на ЭКГ — у 6 (67%), двусторонний синдром запястного канала — у 1 (11%), полиневропатия и хроническая почечная недостаточность — по 4 (44%) случая.
По сравнению с саркомерной ГКМП больные с амилоидозом были достоверно старше (65±9,7 vs 53±14,9 лет, р=0,007), имели более высокий уровень N-концевого промозгового натрийуретического пептида (5623 [ 2674;27345] vs 950 [ 266;2325] пг/мл, р=0,001), чаще демонстрировали снижение фракции выброса ЛЖ <50% (56 vs 3%, р<0,0001), низкий вольтаж (67 vs 4%, р<0,0001) и псевдоинфарктные изменения (44 vs 7%, р=0,004) на ЭКГ. Различий в толщине стенки ЛЖ между группами не было. Для подтверждения диагноза проведено: магнитно-резонансная томография сердца (у 3 больных); биопсия миокарда (у 2), подкожно-жировой клетчатки (у 4), желудочно-кишечного тракта (у 5) и скелетных мышц (у 1); сцинтиграфия миокарда (у 3); иммунохимический анализ крови и мочи (у всех); трепанобиопсия или стернальная пункция (у 5).
Все больные с AL были направлены для специализированного лечения в онкогематологические отделения. 2 из 3 пациентов с ATTRv начали патогенетическую терапию тафамидисом в течение одного месяца после установления диагноза.
Отбор и характеристика включенных в метаанализ выборок для AL и ATTRv представлены на рисунке 1 и в таблице 1. AL зарегистрирован у 32 из 1906 скринированных пациентов, что составило 1,8% (95% доверительный интервал (ДИ): 0,66-3,52). Отмечена высокая гетерогенность исследований (I²=79,88%), однако публикационный сдвиг выявлен не был (рис. 2). Сравнение распространенности AL в российской выборке с данными других популяций не показало статистически значимых различий (р=0,14).
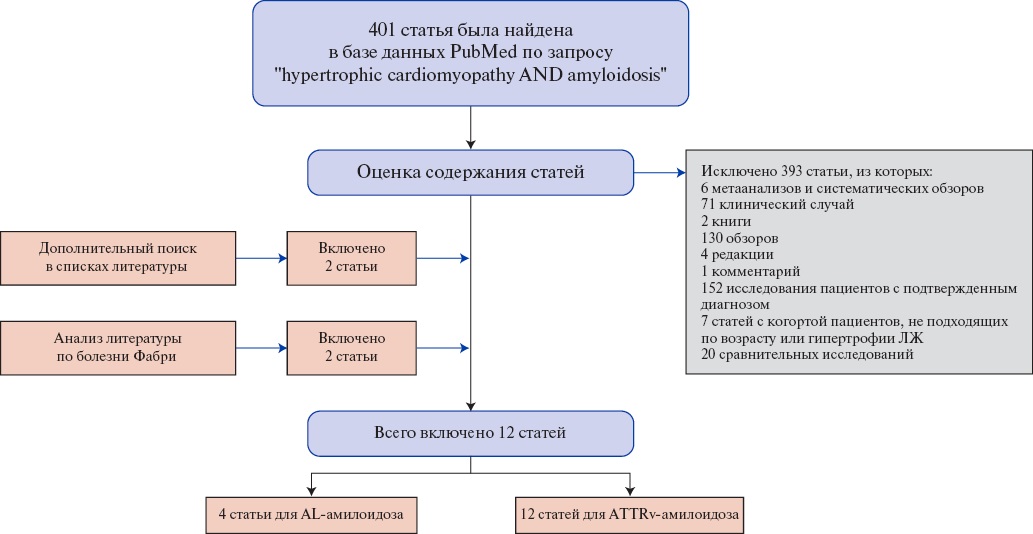
Рис. 1. Поиск публикаций для метаанализа частоты AL и ATTRv сердца среди взрослых больных с фенотипом ГКМП.
Сокращения: ЛЖ — левый желудочек, AL — амилоидоз легких цепей, ATTR — транстиретиновый амилоидоз.
Таблица 1
Публикации, включенные в метаанализ частоты AL, ATTRv и болезни Фабри среди взрослых больных с фенотипом ГКМП
|
Публикации |
Размер выборки, n |
Возраст, лет |
Доля мужчин, % |
Дополнительные критерии отбора |
Доля AL, n (%) |
Доля ATTRv, n (%) |
Доля Фабри, n (%) |
Подтверждение диагноза |
|
Maurizi N, et al., Италия, 2020 [22] |
343 |
60±13 |
58 |
≥40 лет, специализированный центр по амилоидозу |
3 (0,9) |
11 (3,2) |
6 (1,7) |
ДНК, ИМХ, биопсия, сцинтиграфия |
|
Hagege A, et al., Франция, 2019 [23] |
748 |
55±18 |
69 |
регистр ГКМП |
17 (2,3) |
26 (3,5) |
9 (1,2) |
ДНК |
|
Holcman K, et al., Польша, 2024 [9] |
100 |
60±14 |
67 |
нет |
5 (5,0) |
4 (4,0) |
– |
ДНК, ИМХ, биопсия, сцинтиграфия |
|
Gwak SY, et al., Корея, 2024 [8] |
492 |
50±15 |
71 |
исключали несемейные верхушечные ГКМП; специализированный центр по болезни Фабри |
1 (0,2) |
3 (0,6) |
27 (5,5) |
ДНК, МРТ, энзимодиагностика |
|
Lopes LR, et al., Великобритания, 2019 [11] |
770 |
49±16 |
67 |
специализированный центр по кардиомиопатиям |
– |
1 (0,1) |
– |
ДНК, сцинтиграфия |
|
Oktay V, et al., Турция, 2023 [24] |
392 |
50±16 |
68 |
многоцентровое исследование ГКМП |
– |
1 (0,3) |
2 (0,5) |
ДНК |
|
Tran Vu MT, et al., Вьетнам, 2019 [25] |
104 |
49±16 |
64 |
нет |
– |
0 (0) |
1 (1,0) |
ДНК |
|
Jaaskelainen P, et al., Финляндия, 2019 [26] |
382 |
53±14 |
62 |
нет |
– |
0 (0) |
2 (0,5) |
ДНК |
|
Mazzarotto F, et al., Италия, 2018 [27] |
613 |
46±19 |
63 |
специализированный центр по кардиомиопатиям |
– |
6 (1,0) |
4 (0,7) |
ДНК |
|
Hoss S, et al., Канада, 2020 [28] |
1343 |
нет данных |
нет данных |
специализированный центр по ГКМП |
– |
2/691 (0,3) |
11/1148 (1,0) |
ДНК, МРТ, энзимодиагностика |
|
Nguyen K, et al., Франция, 2019 [29] |
200 |
55 |
66 |
нет |
– |
1 (0,5) |
1 (0,5) |
ДНК, энзимодиагностика |
|
Damy Т, et al., Франция, 2016 [10] |
298 |
62±12 |
74 |
специализированный центр по амилоидозу |
– |
15 (5,0) |
– |
ДНК, биопсия, сцинтиграфия |
|
Savostyanov K, et al., Россия, 2022 [4] |
1009 |
50 |
57 |
нет |
– |
– |
4 (0,4) |
ДНК, энзимодиагностика |
|
Barman HA, et al., Турция, 2019 [19] |
80 |
42±13 |
66 |
18-65 лет, необструктивная ГКМП |
– |
– |
2 (2,5) |
ДНК, энзимодиагностика |
|
Azevedo O, et al., Португалия, 2020 [18] |
780 |
66±14 |
60 |
исключение носителей варианта с эффектом основателя |
– |
– |
7 (0,9) |
ДНК, энзимодиагностика |
|
Zemаnek D, et al., Чехия, 2022 [30] |
589 |
58±15 |
66 |
нет |
– |
– |
6 (1,0) |
ДНК, энзимодиагностика |
|
Собственные данные, Россия, 2025 |
223 |
54±15 |
55 |
нет |
6 (2,7) |
3 (1,4) |
1 (0,5) |
ДНК, ИМХ, биопсия, сцинтиграфия, энзимодиагностика |
Сокращения: ГКМП — гипертрофическая кардиомиопатия, ДНК — дезоксирибонуклеиновая кислота (генетический анализ), ИМХ — иммунохимический анализ, МРТ — магнитно-резонансная томография, AL — амилоидоз легких цепей иммуноглобулинов, ATTRv — наследственный транстиретиновый амилоидоз.

Рис. 2. Метаанализ частоты встречаемости AL среди взрослых больных с фенотипом ГКМП. Forest-plot (A) демонстрирует высокую гетерогенность включенных исследований; funnel-plot (Б) указывает на нахождение всех публикаций внутри ДИ.
ATTRv зарегистрирован у 73 из 5356 пациентов, что составило 1,2% (95% ДИ: 0,53-2,15). Гетерогенность исследований оказалась высокой (I²=86,03%), однако публикационный сдвиг выявлен не был (рис. 3). Частота ATTRv в российской когорте не отличалась от других исследований (p=0,69).
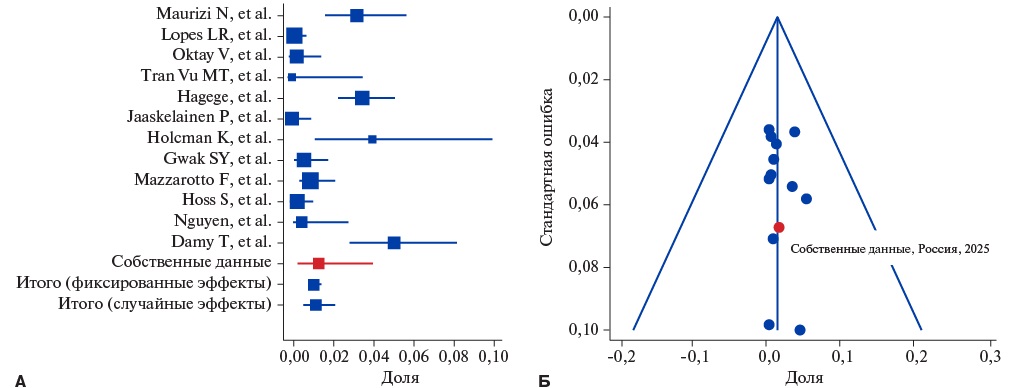
Рис. 3. Метаанализ частоты встречаемости ATTRv среди взрослых больных с фенотипом ГКМП. Forest-plot (A) демонстрирует высокую гетерогенность включенных исследований; funnel-plot (Б) указывает на нахождение всех публикаций внутри ДИ.
У 66-летнего пациента с обструктивной ГКМП после миосептэктомии выявлен патогенный вариант в гене GLA (p.N215S). Сниженная активность α-галактозидазы А (0,66 мкмоль/л/ч, норма >1,89) и повышенный уровень lyso-Gb3 (5,46 нг/мл, норма ≤2,1) в крови подтвердили болезнь Фабри. Гистология миокарда показала гипертрофию кардиомиоцитов с выраженной вакуолизацией. На ЭКГ присутствовал типичный признак — полная блокада правой ножки пучка Гиса. Системных проявлений не было. Ферментозаместительная терапия начата через 2 мес. Семейный скрининг выявил бессимптомное носительство патогенного варианта у дочери (поставлена под диспансерное наблюдение).
Отбор и характеристика включенных в метаанализ исследований для болезни Фабри представлены на рисунке 4 и в таблице 1. Болезнь Фабри зарегистрирована у 1,2% (95% ДИ: 0,72-1,75) — у 83 из 7103 пациентов. Гетерогенность исследований оказалась высокой (I²=73,12%), признаков публикационного сдвига не выявлено (рис. 5). Сравнение нашей группы с данными метаанализа других исследований показало значимое различие (p=0,018). Однако после исключения специализированного по болезни Фабри центра [8], в котором распространенность фенокопии была равной 5,5%, статистических различий не наблюдали (р=0,47).
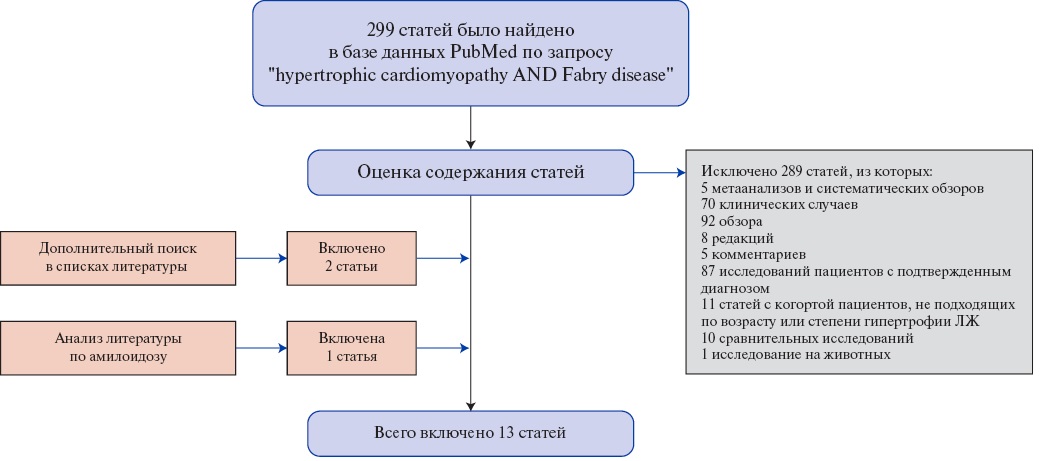
Рис. 4. Поиск публикаций для метаанализа частоты болезни Фабри среди взрослых больных с фенотипом ГКМП.
Сокращение: ЛЖ — левый желудочек.
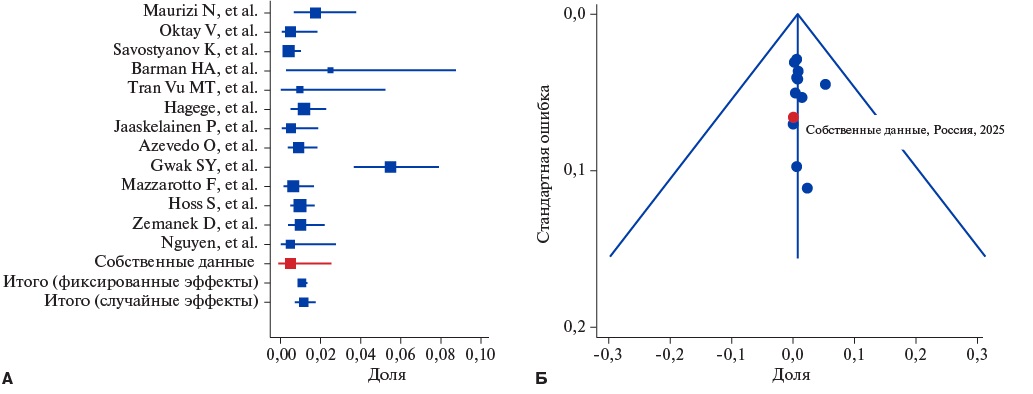
Рис. 5. Метаанализ частоты встречаемости болезни Фабри среди взрослых больных с фенотипом ГКМП. Forest-plot (A) демонстрирует высокую гетерогенность включенных исследований; funnel-plot (Б) указывает на нахождение всех публикаций внутри ДИ.
В нашей выборке частота встречаемости AL и ATTRv составила 2,7% и 1,4%, соответственно, и оказалась сопоставима с данными других исследований. Мы проанализировали публикации, вошедшие в метаанализ, в которых результаты отличались от средних значений. В исследовании Holcman K, et al. (2024) амилоидоз встречался чаще (5% AL, 4% ATTRv), что, вероятно, связано с широким применением биопсии сердца [9]. В работе Damy T, et al. (2016) частота ATTRv достигла 5%, что может быть объяснено высокой долей пациентов с симметричной гипертрофией ЛЖ, тяжелой сердечной недостаточностью и чернокожих, среди которых наблюдается высокая распространенность TTR-варианта p.V122I (V142I) [10]. В исследовании Lopes LP, et al. (2019), напротив, распространенность ATTRv составила всего 0,1%. Из 6 носителей редких вариантов в гене TTR (0,8%) амилоидоз сердца был подтвержден только у 1 пациента. Авторы подчеркивают необходимость клинического подтверждения диагноза у носителей TTR-вариантов из-за неполной пенетрантности и возможного наличия других причин фенотипа, например, мутаций в саркомерных генах [11]. Следует учесть, что сцинтиграфия при некоторых TTR-вариантах дает ложноотрицательные результаты [12].
Мы не исключаем недооценку распространенности ненаследственной формы ATTR (ATTRwt), которая могла быть связана с отсутствием стандартизированной диагностики, в особенности в первые годы включения пациентов, и относительно молодым возрастом наших больных (54 года), в то время как ATTRwt-амилоидоз чаще дебютирует в старческом возрасте [13].
В метаанализе 2022г [14] частота амилоидоза у больных с фенотипом ГКМП составила 7%, включая 4,5% AL, что выше наших 1,8%. Очевидно, это обусловлено включением данных только из двух специализированных центров, в одном из которых включались пациенты 40 лет и старше. В метаанализе Antonopoulos AS, et al. (2022) было рассмотрено 4 исследования [15], частота ATTR варьировала от 2 до 15% в зависимости от дизайна исследования и экспертности центра. Из-за высокой гетерогенности усредненные значения не приводятся, поэтому сопоставить результаты с нашим метаанализом проблематично.
У 1 пациента с обструктивной ГКМП была выявлена болезнь Фабри с поздним дебютом и изолированным кардиальным фенотипом, характерным для обнаруженного патогенного варианта в гене GLA. Единственным "красным флагом" стала блокада правой ножки пучка Гиса, что подчеркивает диагностическое значение ЭКГ [16]. Более половины больных с гипертрофией ЛЖ при болезни Фабри имеют асимметричное утолщение межжелудочковой перегородки, 10% нуждаются в хирургическом лечении обструкции, и четверть не имеют внесердечных проявлений [17], что увеличивает схожесть этой фенокопии с саркомерной ГКМП и затрудняет ее диагностику только на основании клинических признаков. В 2 российских исследованиях, нашем и Savostyanov K, et al. (2022) [4], скрининг проводился в неспециализированных учреждениях, что объясняет низкую (0,4-0,5%), по сравнению со средними данными, частоту заболевания. В то же время в исследовании Gwak SY, et al. распространенность в 5,5% очевидно связана с экспертностью центра [8]. Распространенность также зависела от региона проживания, где могут встречаться варианты с эффектом основателя [18] и от возраста пациентов (чем моложе когорта, тем выше вероятность выявления болезни Фабри) [19].
В метаанализе 26 исследований (>10000 пациентов) частота болезни Фабри среди больных с ГКМП или неясной гипертрофией ЛЖ (≥1,3 см) после переоценки патогенности вариантов в гене GLA по критериям ACMG снизилась с 1,4% до 1,2% [20]. В другом метаанализе (16 исследований, 5491 пациент) частота составила 0,9% и не различалась у мужчин и женщин [21]. В нашем метаанализе частота болезни Фабри среди пациентов с ГКМП составила 1,2% (0,9% без учета данных Gwak SY, et al.), что сопоставимо с ранее опубликованными результатами и очевидно отражает истинную распространенность этой фенокопии.
Ограничения исследования включают небольшую собственную когорту и отсутствие в некоторых публикациях данных о методах верификации диагноза фенокопий, что могло привести к завышению их распространенности.
Фенокопии составляют существенную долю в спектре причин развития фенотипа ГКМП у взрослых. Результаты метаанализа, охватывающего данные из разных регионов мира, включая Россию, можно считать репрезентативными. Повышенная клиническая настороженность в сочетании с обязательным проведением генетического тестирования могут способствовать повышению выявляемости редких заболеваний, маскирующихся под ГКМП.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Бокерия Л.А., Шляхто Е.В., Габрусенко С.А. и др. Гипертрофическая кардиомиопатия. Клинические рекомендации 2025. Российский кардиологический журнал. 2025;30(5):6387. doi:10.15829/1560-4071-2025-6387. EDN: BUUCJT.
2. Благова О.В., Заклязьминская Е.В., Коган Е.А. и др. Синдром первичной гипертрофии миокарда: клинико-морфологическая, генетическая диагностика и сопоставление саркомерных вариантов кардиомиопатии и ее фенокопий. Рациональная Фармакотерапия в Кардиологии. 2019;15(4):484-94. doi:10.20996/1819-6446-2019-15-4-484-494. EDN: RUXARV.
3. Насонова С.Н., Жиров И.В., Шошина А.А. и др. Экспертный центр по амилоидозу сердца: реалии и перспективы. Терапевтический архив. 2024;96(4):321-9. doi:10.26442/00403660.2024.04.202677.
4. Savostyanov K, Pushkov A, Zhanin I, et al. The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: a Russian nationwide screening program using NGS technology. Orphanet J Rare Dis. 2022;17(1):199. doi:10.1186/s13023-022-02319-4.
5. Chumakova OS, Baklanova TN, Milovanova NV, Zateyshchikov DA. Hypertrophic Cardiomyopathy in Underrepresented Populations: Clinical and Genetic Landscape Based on a Russian Single-Center Cohort Study. Genes (Basel). 2023;14:2042. doi:10.3390/genes14112042.
6. Лысенко (Козловская) Л. В., Рамеев В.В., Моисеев С.В. и др. Клинические рекомендации по диагностике и лечению системного амилоидоза. Клиническая фармакология и терапия. 2020;29(1):13-24. doi:10.32756/0869-5490-2020-1-13-24.
7. Brooke BS, Schwartz TA, Pawlik TM. MOOSE Reporting Guidelines for Meta-analyses of Observational Studies. JAMA Surg. 2021;156(8):787-8. doi:10.1001/jamasurg.2021.0522.
8. Gwak SY, Seo J, Seo GH, et al. Role of Genetic Testing in Diagnosis and Prognosis Prediction in Hypertrophic Cardiomyopathy in Korea. J Korean Med Sci. 2024;39(50): e313. doi:10.3346/jkms.2024.39.e313.
9. Holcman K, Kostkiewicz M, Szot W, et al. Transthyretin amyloid cardiomyopathy in patients with unexplained increased left ventricular wall thickness. Int J Cardiovasc Imaging. 2024;40(8):1693-703. doi:10.1007/s10554-024-03158 z.
10. Damy T, Costes B, Hagège AA, et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016;37(23):1826-34. doi:10.1093/eurheartj/ehv583.
11. Lopes LR, Futema M, Akhtar MM, et al. Prevalence of TTR variants detected by wholeexome sequencing in hypertrophic cardiomyopathy. Amyloid. 2019;26(4):243-7. doi:10.1080/13506129.2019.1665996.
12. Rahim MA, Jani V, Gupta V, et al. High rate of false negative 99mTc-pyrophosphate scintigraphy scans in patients with Leu58His transthyretin amyloid cardiomyopathy. Amyloid. 2025;23:1-3. doi:10.1080/13506129.2025.2493688.
13. Полякова А.А., Семернин Е.Н., Ситникова М.Ю. и др. Транстиретиновый амилоидоз в когорте пациентов с хронической сердечной недостаточностью старческого возраста и долгожителей. Кардиология. 2018;58(2S):12-8. doi:10.18087/cardio.2390.
14. Aimo A, Merlo M, Porcari A, et al. Redefining the epidemiology of cardiac amyloidosis. A systematic review and meta-analysis of screening studies. Eur J Heart Fail. 2022;24(12):2342-51. doi:10.1002/ejhf.2532.
15. Antonopoulos AS, Panagiotopoulos I, Kouroutzoglou A, et al. Prevalence and clinical outcomes of transthyretin amyloidosis: a systematic review and meta-analysis. Eur J Heart Fail. 2022;24(9):1677-96. doi:10.1002/ejhf.2589.
16. Чумакова О.С., Исаева М.Ю., Королева О.С. и др. Место электрокардиографии в диагностике кардиомиопатий и спортивного сердца. Российский кардиологический журнал. 2020;25(3S):4023. doi:10.15829/1560-4071-2020-4023. EDN: NRZSRT.
17. Моисеев С.В., Тао Е.А., Моисеев А.С. и др. Болезнь Фабри как причина гипертрофической кардиомиопатии. Клиническая фармакология и терапия. 2023;32(1):36-41. doi:10.32756/0869-5490-2023-1-36-41.
18. Azevedo O, Marques N, Reis L, et al. Predictors of Fabry disease in patients with hypertrophic cardiomyopathy: How to guide the diagnostic strategy? Am Heart J. 2020;226:114- 26. doi:10.1016/j.ahj.2020.04.006.
19. Barman HA, İkitimur B, Kılıçkıran Avcı B, et al. The Prevalence of Fabry Disease Among Turkish Patients with Non-Obstructive Hypertrophic Cardiomyopathy: Insights from a Screening Study. Balkan Med J. 2019;36(6):354-8. doi:10.4274/balkanmedj.galenos.2019.2019.5.125.
20. Monda E, Diana G, Graziani F, et al. Impact of GLA Variant Classification on the Estimated Prevalence of Fabry Disease: A Systematic Review and Meta-Analysis of Screening Studies. Circ Genom Precis Med. 2023;16(6): e004252. doi:10.1161/CIRCGEN.123.004252.
21. Doheny D, Srinivasan R, Pagant S, et al. Fabry Disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J Med Genet. 2018;55(4):261-8. doi:10.1136/jmedgenet-2017-105080.
22. Maurizi N, Rella V, Fumagalli C, et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int J Cardiol. 2020;300:191-5. doi:10.1016/j.ijcard.2019.07.051.
23. Hagège A, Puscas T, El Hachmi M, et al. The French hypertrophic cardiomyopathy gene register: A systematic large gene screening for hypertrophic cardiomyopathy. Int J Cardiol. 2024;417:132542. doi:10.1016/j.ijcard.2024.132542.
24. Oktay V, Tüfekçioğlu O, Yılmaz DÇ, et al. The Definition of Sarcomeric and NonSarcomeric Gene Mutations in Hypertrophic Cardiomyopathy Patients: A Multicenter Diagnostic Study Across Türkiye. Anatol J Cardiol. 2023;27(11):628-38. doi:10.14744/AnatolJCardiol.2023.2805.
25. Tran Vu MT, Nguyen TV, Huynh NV, et al. Presence of Hypertrophic Cardiomyopathy Related Gene Mutations and Clinical Manifestations in Vietnamese Patients With Hypertrophic Cardiomyopathy. Circ J. 2019;83(9):1908-16. doi:10.1253/circj.CJ-19-0190.
26. Jääskeläinen P, Vangipurapu J, Raivo J, et al. Genetic basis and outcome in a nationwide study of Finnish patients with hypertrophic cardiomyopathy. ESC Heart Fail. 2019;6(2):436- 45. doi:10.1002/ehf2.12420.
27. Mazzarotto F, Girolami F, Boschi B, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. 2019;21(2):284-92. doi:10.1038/s41436-018-0046-0.
28. Hoss S, Habib M, Silver J, et al. Genetic Testing for Diagnosis of Hypertrophic Cardiomyopathy Mimics: Yield and Clinical Significance. Circ Genom Precis Med. 2020;13(2): e002748. doi:10.1161/CIRCGEN.119.002748.
29. Nguyen K, Roche S, Donal E, et al. Whole Exome Sequencing Reveals a Large Genetic Heterogeneity and Revisits the Causes of Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2019;12(5): e002500. doi:10.1161/CIRCGEN.119.002500.
30. Zemánek D, Januška J, Honěk T, et al. Nationwide screening of Fabry disease in patients with hypertrophic cardiomyopathy in Czech Republic. ESC Heart Fail. 2022;9(6):4160-6. doi:10.1002/ehf2.14135.
К.м.н., врач-кардиолог ГБУЗ ГКБ №17 ДЗМ, доцент кафедры терапии, кардиологии и функциональной диагностики с курсом нефрологии ФГБУ ДПО ЦГМА УДП РФ
ул. Волынская, д.7, Москва, 119620,
ул. Маршала Тимошенко, д.19, стр. 1А, Москва, 121359
Студент
ул. Трубецкая, д.8, стр. 2, Москва, 119048
Д.м.н., профессор кафедры терапии, кардиологии и функциональной диагностики с курсом нефрологии
ул. Маршала Тимошенко, д.19, стр. 1А, Москва, 121359
Врач-кардиолог
ул. Волынская, д.7, Москва, 119620
Врач функциональной диагностики
ул. Волынская, д.7, Москва, 119620
Врач-терапевт
ул. Волынская, д.7, Москва, 119620
Зав. терапевтическим отделением
ул. Волынская, д.7, Москва, 119620
Зав. отделением функциональной диагностики
ул. Волынская, д.7, Москва, 119620
Зав. отделением кардиологии
ул. Волынская, д.7, Москва, 119620
К.м.н., зам. главного врача
ул. Волынская, д.7, Москва, 119620
Д.м.н., профессор, зав. кафедрой терапии, кардиологии и функциональной диагностики с курсом нефрологии ФГБУ ДПО ЦГМА УДП РФ, заведующий сердечно-сосудистым центром ГБУЗ ГКБ №29 им. Н.Э. Баумана
ул. Волынская, д.7, Москва, 119620,
Госпитальная пл., д.2, Москва, 111020
Чумакова О.С., Кертиева Л.Э., Минушкина Л.О., Ухова Л.В., Алькам Д.М., Алиева З.И., Ильенкова Н.А., Архипкина Т.С., Волошина Н.М., Бакланова Т.Н., Затейщиков Д.А. Распространенность амилоидоза сердца и болезни Фабри среди взрослых больных с фенотипом гипертрофической кардиомиопатии: данные российского одноцентрового исследования и мета-анализ. Российский кардиологический журнал. 2025;30(10):6412. https://doi.org/10.15829/1560-4071-2025-6412. EDN: ZEUPAG
Chumakova O.S., Kertieva L.E., Minishkina L.O., Ukhova L.V., Alkam D.M., Alieva Z.I., Ilyenkova N.A., Arkhipkina T.S., Voloshina N.M., Baklanova T.N., Zateyshchikov D.A. Prevalence of cardiac amyloidosis and Fabry disease among adult patients with hypertrophic cardiomyopathy phenotype: data from a Russian single-center study and meta-analysis. Russian Journal of Cardiology. 2025;30(10):6412. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6412. EDN: ZEUPAG













































