


Содержание
Перейти к:
 М. Г. Бубнова,
М. В. Ежов,
Д. М. Аронов,
А. С. Галявич,
В. С. Гуревич,
Д. В. Дупляков,
В. К. Зафираки,
Н. С. Карамнова,
В. В. Кашталап,
Г. А. Коновалов,
А. Н. Мешков,
А. Г. Обрезан,
А. А. Семенкин,
И. В. Сергиенко,
А. Е. Филиппов
М. Г. Бубнова,
М. В. Ежов,
Д. М. Аронов,
А. С. Галявич,
В. С. Гуревич,
Д. В. Дупляков,
В. К. Зафираки,
Н. С. Карамнова,
В. В. Кашталап,
Г. А. Коновалов,
А. Н. Мешков,
А. Г. Обрезан,
А. А. Семенкин,
И. В. Сергиенко,
А. Е. Филиппов https://doi.org/10.15829/1560-4071-2025-6364
EDN: VNFGFQ
Перейти к:
Гипертриглицеридемия (триглицерид-богатые липопротеины и их ремнанты): роль в развитии атеросклеротических сердечно-сосудистых заболеваний и стратегия контроля. Заключение Комитета экспертов Российского кардиологического общества (РКО), Национального общества по изучению атеросклероза (НОА), Российского общества кардиосоматической реабилитации и вторичной профилактики (РосОКР)
Бубнова М.Г., Ежов М.В., Аронов Д.М., Галявич А.С., Гуревич В.С., Дупляков Д.В., Зафираки В.К., Карамнова Н.С., Кашталап В.В., Коновалов Г.А., Мешков А.Н., Обрезан А.Г., Семенкин А.А., Сергиенко И.В., Филиппов А.Е. Гипертриглицеридемия (триглицерид-богатые липопротеины и их ремнанты): роль в развитии атеросклеротических сердечно-сосудистых заболеваний и стратегия контроля. Заключение Комитета экспертов Российского кардиологического общества (РКО), Национального общества по изучению атеросклероза (НОА), Российского общества кардиосоматической реабилитации и вторичной профилактики (РосОКР). Российский кардиологический журнал. 2025;30(5):6364. https://doi.org/10.15829/1560-4071-2025-6364. EDN: VNFGFQ
Bubnova M.G., Ezhov M.N., Aronov D.M., Galyavich A.S., Gurevich V.S., Duplyakov D.V., Zafiraki V.K., Karamnova N.S., Kashtalap V.V., Konovalov G.A., Meshkov A.N., Obrezan A.G., Semenkin A.A., Sergienko I.V., Filippov A.E. Hypertriglyceridemia (triglyceride-rich lipoproteins and their remnants): role in the development of atherosclerotic cardiovascular diseases and control strategy. Opinion of the Expert Committee of the Russian Society of Cardiology, the National Atherosclerosis Society, and the Russian Society of Cardiac and Somatic Rehabilitation and Secondary Prevention. Russian Journal of Cardiology. 2025;30(5):6364. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6364. EDN: VNFGFQ
Смертность от сердечно-сосудистых заболеваний (ССЗ) значимо опережает смертность от инфекционных и онкологических болезней. По данным Всемирной организации здравоохранения к 2030г от ССЗ ежегодно будут умирать ~23,6 млн человек1. Объем доказательств, накопленный к настоящему времени, свидетельствует о том, что повышенный уровень холестерина (ХС) липопротеинов (ЛП) низкой плотности (ЛНП) — неоспоримый фактор развития атеросклеротических ССЗ (АССЗ) и его снижение — важная задача в рамках первичной и вторичной профилактики [1-3]. Однако причины и генез атеросклеротической болезни не всегда можно объяснить с позиции нарушения липидного обмена, основанного исключительно на ЛНП.
В последние годы наблюдается рост количества исследований, посвященных гипертриглицеридемии (ГТГ). Обсуждаются механизмы формирования ГТГ, определяющие ее значимость в атерогенезе. В этой связи возникает необходимость в актуализации ключевых вопросов, определяющих первопричину развития АССЗ при ГТГ: это молекулы триглицеридов (ТГ) сами по себе, ТГ-богатые ЛП или их ремнанты (остатки). Во-вторых, не ясно: какие компоненты ТГ-богатых ЛП и/или их ремнантов связаны с повышенным риском развития АССЗ и достаточно ли они изучены, чтобы использовать их в качестве терапевтических мишеней? Можно выделить несколько главных аргументов, обосновывающих возрастание научного интереса к проблеме ГТГ и пересмотру роли ТГ (ТГ-богатых ЛП) в развитии АССЗ:
— завершение генетических исследований (с менделевской рандомизацией), подтвердивших атерогенную природу ТГ-богатых ЛП, через которые опосредуется связь ГТГ с риском развития АССЗ;
— получение доказательств из эпидемиологических исследований о роли ГТГ в развитии АССЗ независимо от уровня ХС ЛНП;
— сохранение существенного остаточного риска развития АССЗ (и их осложнений) у пациентов, в т.ч. достигших целевых (низких) значений ХС ЛНП на ХС-снижающей терапии;
— получение доказательств значимой пользы в снижении сердечно-сосудистого риска (ССР) с помощью подхода, основанного на контроле уровня ТГ в крови.
На современном этапе существует необходимость в экспертных рекомендациях по ведению пациентов с ГТГ, выработке согласованной позиции по проблеме резидуального (остаточного) ССР, немедикаментозному вмешательству в образ жизни и применению лекарственной терапии для коррекции повышенного уровня ТГ и снижения риска развития АССЗ. Особого обсуждения требует ведение пациентов с ГТГ высокого/очень высокого ССР в первичной профилактике, с доказанными АССЗ (во вторичной профилактике), сахарным диабетом (СД), хронической болезнью почек (ХБП), тяжелой ГТГ.
Согласованные позиции экспертов, изложенные в представленном документе, основаны на анализе и обсуждении имеющихся научных доказательствах. Документ направлен на расширение знаний практикующих врачей в области липидологии, повышение их осведомленности о роли ГТГ в патогенезе атеросклероза и важности оценки уровня ТГ, устранение определенного пробела в клинической практике по ведению пациентов с разной степенью выраженности ГТГ.
В российских и европейских рекомендациях, посвященных нарушениям липидного обмена, определение уровня ТГ в плазме/сыворотке крови рекомендуется всем пациентам как часть рутинного анализа липидного спектра крови (класс рекомендаций I) [1][2]. Концентрация ТГ в плазме крови отражает концентрацию так называемых ТГ-богатых ЛП, которые в состоянии натощак представлены ЛП очень низкой плотности (ЛОНП) [4]. В состоянии не натощак (в период от 1 до 6 ч после последнего приема обычной пищи) у индивидуумов из общей популяции содержание ТГ в плазме крови повышается в среднем на ~0,3 ммоль/л (26 мг/дл) [5][6]. На повышение концентрации ТГ в плазме крови не натощак в разной степени влияют ТГ-богатые ЛП, образующиеся после еды, — это хиломикроны (ХМ) и их ремнанты, а также синтезирующиеся в печени ЛОНП и их ремнанты (ремнанты мелких ЛОНП2 по существу есть ЛП промежуточной плотности (ЛПП)) [7].
В таблице 1 представлена классификация ГТГ по степени ее выраженности согласно консенсусу Европейского общества атеросклероза, принятому в 2021г [8]. Хотя обозначенные пороговые значения ТГ являются условными, но они согласуются с современными знаниями и имеют определенное значение для клинической практики. Оптимальный уровень ТГ <1,2 ммоль/л отражает эффективный липолиз ТГ-богатых ЛП и низкую концентрацию их ремнантов в плазме крови, он ассоциирован с низким ССР [2]. Концентрация ТГ >1,7 ммоль/л определяется клинически значимой для развития АССЗ у мужчин и женщин (по данным крупнейшего эпидемиологического Фрамингемского исследования) [9].
Таблица 1
Классификация уровней ТГ в плазме (сыворотке) крови
|
Классификация уровней ТГ натощак [адаптировано из 8] |
Уровни ТГ, ммоль/л |
|
Оптимальный |
<1,2 |
|
Пограничный |
1,2-1,7 |
|
Умеренно повышенный (ГТГ) |
1,7-5,7 |
|
Высокий (выраженная ГТГ) |
5,7-10,0 |
|
Экстремально высокий |
>10,0 |
|
Классификация уровней ТГ не натощак [адаптировано из 6] |
|
|
Повышенный (ГТГ) |
≥2,0 |
Сокращения: ГТГ — гипертриглицеридемия, ТГ — триглицериды.
Высокие уровни ТГ (особенно >10,0 ммоль/л) — фактор развития острого панкреатита. При концентрации ТГ от 10 до 20 ммоль/л панкреатит развивается у 3% пациентов и ТГ >20 ммоль/л — у 15%. В некоторых выбранных группах регистрировали и более высокие показатели распространённости острого панкреатита: в немецкой когорте 19% пациентов с уровнем ТГ >11,3 ммоль/л имели панкреатит [10][11]. Каждое повышение содержания ТГ в плазме крови на 1,1 ммоль/л увеличивает риск развития острого панкреатита на 4% [12]. Уровень ТГ в плазме крови, при котором может развиться острый панкреатит, варьирует особенно у пациентов, предрасположенных к нему, и у лиц с эпизодами острого панкреатита в анамнезе [13].
К оценке уровня ТГ в сыворотке крови не натощак (после еды) стали проявлять большое внимание в последнее десятилетие, поскольку этот показатель может лучше информировать о среднем содержании ТГ в крови в течение дня у конкретного индивидуума и служить доказательством нарушений транспорта липидов как за счет ЛП кишечного, так и ЛП печеночного происхождения. Концентрацию ТГ в плазме крови не натощак ≥2,0 ммоль/л принято считать отрезной точкой, определяющей постпрандиальный подъем ТГ как фактор ССР [6][14].
ГТГ предлагается рассматривать в качестве маркера высокого содержания ХС в ТГ-богатых ЛП (ряд специалистов применяет термин "ХС ремнантов") [15]. Исключение составляют лица с экстремальной ГТГ на фоне полного дефицита липопротеинлипазы (ЛПЛ), который встречается у одного человека из миллиона. Существуют определенные технические сложности и ограничения в аналитических подходах к точному измерению уровня ТГ-богатых ЛП и содержащегося в них ХС. Это связано с нестабильностью динамического катаболизма данных ЛП и изменчивостью их липидного и белкового состава [16]. При ГТГ концентрацию "ХС ремнантов" предлагается рассчитывать по формуле [17]:
ХС ремнантов = общий ХС – ХС ЛНП – ХС ЛП высокой плотности (ЛВП).
На повышенный уровень "ХС ремнантов" указывает значение натощак ≥0,8 ммоль/л (≥30 мг/дл) и в условиях не натощак ≥0,9 ммоль/л (≥35 мг/дл) (по данным эпидемиологических исследований) [6][18]. Постпрандиальная (после приема пищи) концентрация ТГ в плазме крови в полной степени информирует об уровне ремнантов ТГ-богатых ЛП и содержании в них ХС. Постпрандиальная ГТГ более значимо связана с риском развития АССЗ, чем уровень ТГ натощак [17].
Пациентам с ГТГ, страдающим СД, ожирением, метаболическим синдромом или имеющим очень низкий уровень ХС ЛНП для более точной оценки ССР рекомендуется рассчитывать уровень ХС не-ЛВП (класс рекомендаций I) [1][2]. Уровень ХС не-ЛВП рекомендуется определять и пациентам высокого/очень высокого ССР (класс рекомендаций IIа) [1][2]. Установлено, что показатель ХС не-ЛВП в качестве прогностического фактора развития АССЗ превосходит ХС ЛНП [15][19]. ХС не-ЛВП представляет собой суммарную концентрацию всех аполипопротеин (апо)В-содержащих ЛП, включая ТГ-богатые ЛП, ЛНП и ЛП(а), в плазме/сыворотке крови как натощак, так и после приема пищи. ХС не-ЛВП рассчитывается по формуле:
ХС не-ЛВП = общий ХС – ХС ЛВП.
В настоящее время идет поиск показателей и методов, способных более полно отражать независимый атерогенный потенциал ТГ-богатых ЛП/ремнантов, их структурную и метаболическую изменчивость, количественный состав после приема пищи. Учитывая доказанную высокую атеротромбогенность ТГ-богатых ЛП/ремнантов, определение их уровня в плазме крови может оказаться более информативным, чем предполагалось ранее [16].
По общепризнанной классификации ЛП плазмы крови, разработанной Д. Фредриксоном и состоящей из 5 классов гиперлипидемии (ГЛП), нарушения в виде ГТГ в той или иной степени характерны для ГЛП I, IIb, III, IV и V типов (табл. 2) [20]. ГЛП I типа характеризуется высокой гиперхиломикронемией; ГЛП IIb и IV типов — повышенным образованием в печени ЛОНП, их замедленным катаболизмом или сочетанием того и другого механизма; ГЛП III типа — дефектом захвата и последующим накоплением в плазме крови аномальных ремнантов ХМ и ЛОНП (называемых "β-ЛОНП"), а также ЛПП.
Таблица 2
Классификация ГЛП по фенотипам (по Д. Фредриксону с учетом генетической природы ЛП)
|
Фенотип ГЛП (Фредриксон)/название |
Повышенный уровень ЛП |
Уровень ХС |
Уровень ТГ |
Генетическая природа |
|
I Семейная хиломикронемия |
ХМ |
Норма |
↑↑↑↑ |
Моногенная ГТГ; аутосомно-рецессивный тип, вызывается биаллельными патогенными вариантами генов: LPL, APOC2, APOA5, LMF1, GPIHBP1 или GPD1; проявляется в детстве, раннем юношестве |
|
IIa Семейная ГХС (гипербета-липопротеинемия) |
ЛНП |
↑↑ |
Норма |
Моногенная ГХС; аутосомно-кодоминантный тип, гетерозиготные или биаллельные патогенные варианты генов LDLR, APOB или PCSK9; аутосомно-рецессивный тип вызывается биаллельными патогенными вариантами гена LDLRAP1 |
|
IIb Комбинированная ГЛП (гипербета- и пребета-липопротеинемия) |
ЛНП и ЛОНП |
↑↑ |
↑↑ |
Обычно полигенная ГХС; высокие значения ШГР ХС ЛНП. В сочетании с полигенной ГТГ; высокие значения ШГР ГТГ; и/или редкие гетерозиготные варианты генов LPL, APOC2, APOA5, LMF1, GPIHBP1 или GPD1 |
|
III Семейная дисбета-липопротеинемия (флотирующие беталипопротеины) |
Ремнанты, ЛПП |
↑↑ |
↑↑↑ |
Высокие значения ШГР по ГТГ. Гомозиготность по ε2ε2 генотип АРОЕ или редкие гетерозиготные варианты АРОЕ |
|
IV Семейная ГТГ (гиперпребета-липопротеинемия) |
ЛОНП |
Норма или ↑ |
↑↑ |
Полигенная ГТГ; высокие значения ШГР ГТГ; и/или редкие гетерозиготные варианты генов LPL, APOC2, APOA5, LMF1, GPIHBP1 или GPD1 |
|
V Многофакторная хиломикронемия (хиломикронемия и гиперпребета-липопротеинемия) |
ХМ и ЛОНП |
Норма или ↑ |
↑↑↑↑ |
Полигенная ГТГ; высокие значения ШГР ГТГ; и/или редкие гетерозиготные варианты генов LPL, APOC2, APOA5, LMF1, GPIHBP1 или GPD1 с большим вкладом риск-аллелей, чем при типе IV |
Сокращения: ГЛП — гиперлипидемия, ГТГ — гипертриглицеридемия, ГХС — гиперхолестеринемия, ЛНП — липопротеины низкой плотности, ЛОНП — липопротеины очень низкой плотности, ЛП — липопротеины, ЛПП — липопротеины промежуточной плотности, ТГ — триглицериды, ХМ — хиломикроны, ХС — холестерин, ШГР — шкала полигенного генетического риска.
Учитывая новые данные генетических исследований, представления о генетической первооснове фенотипов ГТГ пересматриваются (табл. 2) [4]. Так, среди пациентов с уровнем ТГ >10 ммоль/л высокая встречаемость ГТГ с моногенной основой и ГТГ V типа с олиго- или полигенной природой; ГЛП IIb типа обычно имеет полигенный характер, как и ГТГ IV типа, но при ГЛП IIb типа есть и другие генетические нарушения в отношении ЛНП, попадающие в другую шкалу [21].
Ключевые положения
ГТГ (уровень ТГ ≥1,7 ммоль/л) — это актуальная проблема для клинической практики. В мире примерно каждый десятый взрослый имеет повышенный уровень ТГ (с возможными вариациями между регионами) [22]. В Копенгагенском исследовании (Copenhagen General Population Study, n=84177) у 27% взрослых общей популяции уровень ТГ находился в пределах от 2 до 10 ммоль/л, а у 0,1% взрослых — >10 ммоль/л [7]. Высокий (>1 ммоль/л) уровень ХС ремнантов ТГ-богатых ЛП встречался у 21% взрослых и находился в пределах от 0,5 до 1,0 ммоль/л у 45%.
В американском исследовании NHANES (National Health and Nutrition Examination Survey) 31% взрослых имели уровень ТГ ≥1,7 ммоль/л, 16,2% — ≥2,3 ммоль/л и 1,1% — ≥5,7 ммоль/л [23]. По данным разных исследований экстремально высокий уровень ТГ (>10 ммоль/л) встречается от 0,1 до 1%, а уровень ТГ >20 ммоль/л реже (примерно у 0,014%) [22][24][25]. Среди 22063 пациентов из Европы и Канады, получающих статины, уровень ТГ ≥1,7 ммоль/л выявлялась у 38,8%, при этом у 35,3% пациентов низкого ССР, 38,5% пациентов с ССЗ и 44,5% пациентов с СД [26].
По данным российского эпидемиологического исследования ЭССЕ-РФ-3 (2020-2022гг, возраст 35-74 лет, n=28399), повышенный (≥1,7 ммоль/л) уровень ТГ имели 32,2% взрослых, причем чаще мужчины (37,3%), чем женщины (28%) [27]. Распространенность ГТГ увеличивалась при сопутствующих заболеваниях и составляла: при артериальной гипертонии — 39,6%, инфаркте миокарда (ИМ) в анамнезе — 37,3%, инсульте в анамнезе — 38,7%, ишемической болезни сердца (ИБС) — 38,1%, ожирении — 44,7% и СД — 52,1%.
По данным ретроспективного поперечного исследования PROMETHEUS (Prevalence of mixed dyslipidemia and severe hypertriglyceridemia in the Russian population), в котором использовалась база результатов определения липидного профиля у 357073 лиц из 254 городов России, уровень ТГ ≥1,7 ммоль/л имелся у 1/3 части населения России (29,2%) [28]. При этом у 16,2% лиц уровень ТГ был умеренно повышенным (≥1,7 — <2,3 ммоль/л), у 12,9% — высоким (≥2,3 — <5,6 ммоль/л), у 0,11% — очень высоким (≥5,6 ммоль/л) и у 0,011% — экстремально высоким (≥10,0 ммоль/л). Частота ГТГ увеличивалась с возрастом и чаще (в 1,23 раза) фиксировалась у мужчин, чем у женщин. Высокая распространенность ГТГ встречалась у мужчин в возрасте 40-49 лет (42,8%) и у женщин — в возрасте 60-69 лет (34,4%).
ГТГ часто сочетается с низкой концентрацией ХС ЛВП в плазме крови, что трансформируется в атерогенную дислипидемию. Дислипидемия — распространенное состояние и ее значимость обычно недооценена практикующими врачами. Примерно у 45% мужчин и 30% женщин, участвовавших в Копенгагенском исследовании, повышенный уровень ТГ (≥1,7 ммоль/л) сочетался с низким ХС ЛВП (<1,0 ммоль/л) [7]. По данным исследования NHANES, дислипидемия отмечалась у 55% пациентов с ССЗ, 62% пациентов с СД и 87% пациентов с метаболическим синдромом [29]. Как видно, наибольшая распространенность атерогенной дислипидемии выявлялась среди лиц с СД и метаболическим синдромом.
Выделяют первичную и вторичную ГТГ. Среди первичных причин повышения уровня ТГ следует упомянуть наследственные механизмы (табл. 2) [4]. Тяжелая ГТГ (с уровнем ТГ >10 ммоль/л) развивается при ряде наследственных нарушений:
1) моногенной хиломикронемии (ГЛП I типа по Д. Фредриксону или синдром семейной хиломикронемии) из-за отсутствия или значительного снижения активности основного фермента гидролиза ТГ-богатых ЛП — ЛПЛ (мутации гена LPL) и факторов функционирования/активности ЛПЛ, таких как: ко-активатор апоCII (мутации гена APOC2) и апоА-V (мутации гена APOA5), фактор 1 созревания липазы (мутация гена LMF1) и гликозилфосфатидилинозитол-прикрепленный белок 1 типа, связывающий ЛВП (GPIHBP1) (мутации гена GPIHBP1); проявляется накоплением ХМ в плазме крови (заболевание проявляется уже в детстве);
2) многофакторной или полигенной хиломикронемии (ГЛП V типа по Д. Фредриксону, смешанная ГТГ) из-за редких гетерозиготных вариантов генов с большим эффектом в генах, идентичных таковым при моногенной хиломикронемии (LPL, APOC2, APOA5, LMF1, GPIHBP1 или GPD1) и/или при многочисленных вариантах генов с малым эффектом в большом числе различных генов (например, APOA1-C3-A4-А5, TRIB, MLXIPL, GCKR, FADS1-2-3, NCAN, APOB, PLTP, ANGPTL3 и т.п.); проявляется накоплением ХМ и ЛОНП в плазме крови.
Умеренная — выраженная ГТГ (с уровнем ТГ 2,0-9,9 ммоль/л) наследственного характера представлена при:
1) многофакторной или полигенной ГТГ (ГЛП IV типа по Д. Фредриксону или семейная ГТГ), развивающейся по причине генетических вариантов генов, сходных со спектром многофакторной хиломикронемии; проявляется накоплением ЛОНП в плазме крови;
2) семейной комбинированной ГЛП (ранее ГЛП IIb типа по Д. Фредриксону), имеющей полигенный характер, с вовлечением генов, регулирующих метаболизм ЛОНП и ЛНП; проявляется накоплением ЛОНП и ЛНП в плазме крови;
3) дисбеталипопротеинемии (ГЛП типа III по Д. Фредриксону, ремнантная ГЛП) из-за определенных полиморфизмов апоЕ — большинство это гомозиготы генотипа ε2/ε2 гена АРОЕ, редко встречаются гетерозиготы по доминантным мутациям АРОЕ; специфическая особенность — электрофоретическая β-подвижность ЛП, выделенных методом препаративного ультрацентрифугирования во фракции с d<1,006 (по плотности как ЛОНП); в результате нарушается опосредованной апоВ, Е-рецептором элиминации ремнантов ХМ/ЛОНП, обогащенных изоформой апоЕ2 и ХС, что приводит к их накоплению в плазме крови (ЛП частицы, имеющие апоЕ2, практически не обладают сродством к апоВ, Е-рецептору).
Моногенные формы ГТГ встречаются редко. В большинстве случаев ГТГ представляет собой полигенное нарушение липидного обмена с заметным влиянием других факторов [4]. Например, для развития генетически обусловленной ГЛП III типа необходимо присутствие других патологических состояний (СД 2 типа, ожирения, гипотиреоза и др.). Наличие сопутствующих заболеваний (ожирение, СД, гипотиреоз, уремия, злоупотребление алкоголем, ХБП и т.д.) может усиливать выраженность ГТГ при ГЛП IV и V типов по Д. Фредриксону.
У пациентов с моногенной хиломикронемией, сопровождающейся высокой концентрацией ХМ в плазме крови, заметно возрастает риск развития острого панкреатита (до 60-70% vs 5-10% при многофакторной хиломикронемии) и отмечается устойчивость к препаратам, снижающим ТГ. Поскольку связанный с ГТГ панкреатит может приводить к летальному исходу, важно понимать, какие препараты и состояния в сочетании с генетической предрасположенностью, повышают вероятность развития этого заболевания. Клинически тяжелая хиломикронемия клинически проявляется также развитием липемии сетчатки, эруптивных ксантом и рецидивирующими болями в животе [30]. Из клинических особенностей, например, дисбеталипопротеинемии III типа следует отметить липоидную дугу роговицы, ксантелазмы, локтевые и коленные ксантомы, ксантомы ладонных линий или пальмарные ксантомы.
Наследственную дисбеталипопротеинемию III типа относят к нарушениям липидного обмена с высокой атерогенностью, тогда как при семейной ГЛП I типа (хиломикронемии) риск развития атеросклероза не увеличивается, несмотря на тяжелую ГТГ [7][31]. Причина в том, что ХМ слишком велики для проникновения через эндотелиальный барьер [32]. К наиболее распространенному типу первичного нарушения липидного обмена относят комбинированную ГЛП (IIb типа), которая составляет до 40% случаев всех ГЛП.
В клинической практике врача чаще встречается вторичная ГТГ, обусловленная негенетическими причинами. Практикующему врачу важно знать эти причины для своевременного выявления и коррекции ГТГ. Факторы, содействующие развитию ГТГ, необходимо свести к минимуму. Следует помнить о контроле уровня ТГ в плазме крови при применении лекарственных средств, повышающих его, и в случае необходимости использовать альтернативные методы. Выделяют следующие причины вторичной ГТГ [14]:
— заболевания: плохо контролируемый СД, ХБП, нефротический синдром, неконтролируемый гипотиреоз, синдром Кушинга, болезни накопления гликогена, острый гепатит, ревматоидный артрит, псориаз, системная красная волчанка, множественная миелома, сепсис и др.;
— образ жизни/нарушение обмена веществ: злоупотребление алкоголем, диета с высоким содержанием насыщенных жиров, углеводов, потребление продуктов с высоким гликемическим индексом, малоподвижный образ жизни, избыточный вес или ожирение, метаболический синдром/инсулинорезистентность, беременность (в третьем триместре уровень ТГ достигает максимума);
— лекарственные средства: некардиоселективные бета-адреноблокаторы, тиазидные диуретики, секвестранты желчных кислот, глюкокортикостероиды, анаболические стероиды, пероральные эстрогены (ралоксифен, тамоксифен, кломифена цитрат, эстрадиол, этинил эстрадиол, конъюгированные эстрогены), L-аспарагиназа, бексаротин, циклофосфамид, ингибиторы протеазы, нейролептики (оланзапин, миртазапин, клозапин), иммунодепрессивные средства (такролимус, сиролимус, циклоспорин, интерфероны).
Вторичные причины ГТГ могут выступать в роли факторов, демаскирующих генетическую предрасположенность и способствующих проявлению определенного фенотипа ГЛП.
Содержание ТГ в плазме крови может колебаться от 0,33 до 120 ммоль/л, что отражает вариабельность скорости секреции и выведения из кровотока ТГ-богатых ЛП [7, 8]. Известно, что даже при уровне ТГ в плазме крови натощак, равном 1,2 ммоль/л, может наблюдаться ряд метаболических нарушений, связанных с накоплением ТГ-богатых ЛП и их ремнантов. При росте содержания ТГ в крови увеличивается и количество этих ЛП-частиц. Условно можно выделить ряд первичных механизмов формирования ГТГ:
— при повышении уровня ТГ до пограничных/умеренных значений чаще преобладает гиперпродукция ЛОНП в печени;
— при высоком уровне ТГ приоритет имеет нарушенный липолиз и клиренс ТГ-богатых ЛП с накоплением ЛОНП, ХМ и их ремнантов в плазме крови;
— при тяжелой ГТГ доминирующей аномалией является низкая скорость клиренса ТГ-богатых ЛП (у пациентов с моногенной ГТГ продукция ТГ-богатых ЛП может быть в нормальных пределах).
Ключевые положения
Основная функция ТГ-богатых ЛП заключается в транспортировке ТГ в жировую ткань для хранения, а также в скелетные и сердечную мышцы для выработки энергии.
ТГ (порядка 85%) поступают в организм в составе пищевого жира и представляют собой сложные эфиры трехатомного спирта глицерина с тремя высшими жирными кислотами (ЖК), содержащими от 12 до 24 углеродных атомов [8]. Часть молекул ТГ в желудке подвергается гидролизу с участием фермента желудочной липазы, но большая часть расщепляется в верхних отделах тонкого кишечника при участии панкреатической липазы. Конечные продукты гидролиза ТГ (ЖК и β-моноглицерид) включаются в мембраны энтероцитов слизистой ворсинок тонкого кишечника, затем они переходят в их цитоплазму и в гладком эндоплазматическом ретикулуме включаются в ресинтез ТГ. В энтероцитах ТГ участвуют в образовании ХМ.
ХМ — самые крупные ЛП частицы (с диаметром от 100 до 1000 нм), содержащие структурный апобелок В48 и образующиеся в большом количестве в энтероцитах при поступлении в организм с пищей ТГ (содержащих длинноцепочечные ЖК), т.е. в постпрандиальную стадию (рис. 1). ХМ являются основной транспортной формой экзогенных (пищевых) липидов (главным образом, ТГ до 95%), они переносят до 70-90 г ТГ в день (~100 ммоль) [31]. ХМ, как и другие ЛП, также переносят фосфолипиды и этерифицированный ХС.
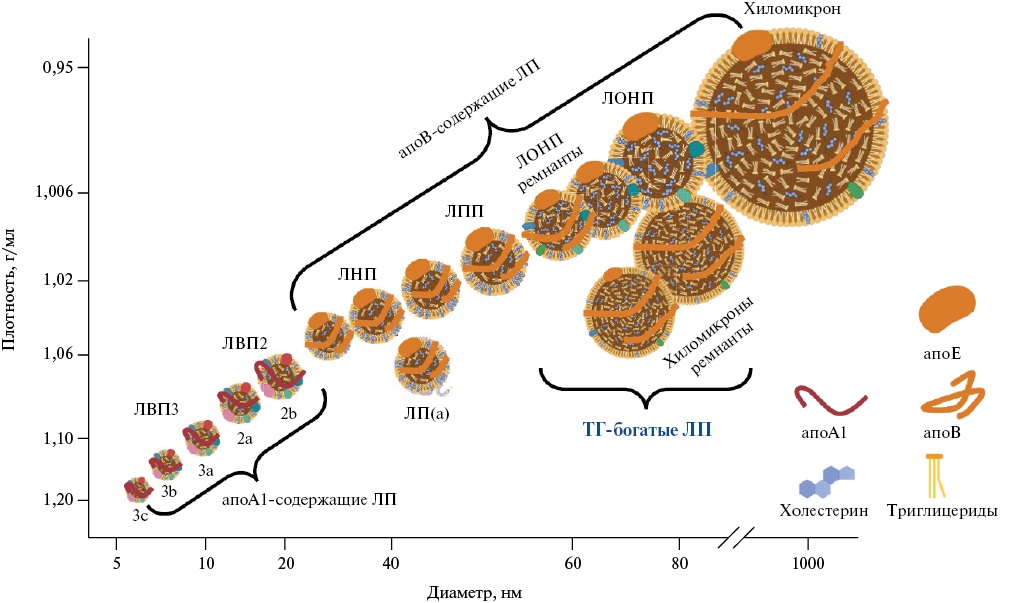
Рис. 1. ЛП: размеры и плотность [адаптирована из 8].
Сокращения: апо — аполипопротеин, ЛВП — липопротеины высокой плотности, ЛНП — липопротеины низкой плотности, ЛОНП — липопротеины очень низкой плотности, ЛП — липопротеины, ЛПП — липопротеины промежуточной плотности, ТГ — триглицериды.
ХМ из энтероцитов поступают в мезентериальные лимфатические сосуды, далее через грудной проток в кровь и с нею транспортируются в периферические капилляры, на поверхности эндотелия которых находятся молекулы периферической ЛПЛ. Этот фермент гидролизует ХМ (в их ядре происходит расщепление до 75% ТГ) [8][33]. Активность ЛПЛ поддерживают ее кофакторы — апоСII и апоАV. В противоположность этому, апоСIII выступает в роли ингибитора ЛПЛ и рассматривается как фактор, влияющий на скорость выведения ТГ-богатых ЛП из кровотока. Низкий уровень апоCIII в крови сопряжен с низким уровнем ТГ, тогда как повышенный уровень, обычно наблюдаемый при инсулинорезистентности и ожирении, с ГТГ. Ангиопоэтиноподобные белки 3, 4 и 8 (ANGPTL 3, 4, 8) подавляют активность ЛПЛ в тканях (жировой ткани, скелетных мышцах, сердце) [34].
На фоне роста концентрации ХМ в плазме крови в печени усиливается образование других ТГ-богатых ЛП — ЛОНП [8]. По структуре и составу ЛОНП сходны с ХМ, но обладают меньшим размером (диаметр варьирует от 25 до 100 нм), содержат меньше ТГ (50-70% от массы частицы), но больше ХС, фосфолипидов и имеют в своем составе структурный апоВ100. ЛОНП синтезируются в печени и являются транспортной формой эндогенных ТГ. ЛОНП переносятся 20-30 г ТГ в день [31].
ЛОНП отличаются чрезвычайной гетерогенностью как по размерам частиц, так и по их составу: апоВ и апоЕ находятся в различном конформационном состоянии в больших ЛОНП1 (Sf 60-400) и мелких ЛОНП2 (Sf 20-60) частицах, что определяет их субстратное сродство к липолитическим ферментам и степень взаимодействия со специфическими рецепторами клеточных мембран. ХМ и ЛОНП1 конкурируют за один и тот же механизм липолиза с участием периферической ЛПЛ. ЛОНП подвергаются гидролизу с образованием ремнантов ЛОНП и ЛПП. В результате последовательного действия периферической ЛПЛ и печеночной липазы ЛОНП и ЛПП превращаются в частицы ЛНП. В условиях повышенного печеночного липогенеза, приводящего к гиперпродукции ЛОНП, процесс превращения ЛОНП в ЛНП замедляется. Это дополнительно содействует аккумуляции ТГ-богатых ЛП и их ремнантов в плазме крови. Связывание частицы ЛНП с апоВ, Е-рецепторами (классическими рецепторами к ЛНП) опосредуется молекулой апоВ100, а связывание ремнантов ТГ-богатых ЛП — апоЕ [33].
Во время липолиза размер ТГ-богатых ЛП уменьшается на фоне одновременного снижения содержания ТГ в их ядре и увеличения концентрации ХС за счет обмена ТГ из частиц ТГ-богатых ЛП на эфиры ХС из частиц ЛНП и ЛВП при участии белка-переносчика эфиров ХС. При этом в составе ТГ-богатых ЛП сохраняются их апобелки — апоВ48, либо апоВ100. Ремнанты ТГ-богатых ЛП имеют высокое соотношение эфиров ХС к ТГ и повышенный уровень апоЕ. Присутствие молекул апоЕ требуется для быстрого рецептор-опосредованного удаления ремнантных частиц из кровотока. Исчезновение ремнантов ХМ из плазмы крови осуществляется через специфический белок-1, подобный рецептору ЛНП, (LRP1 — receptor related protein1). Основной белок ХМ — апоВ48 из-за особенностей структуры не способен связываться с ЛНП-рецепторами (домен, ответственный за связывание апоВ, Е-рецепторами, расположен на С-конце молекулы апоВ100) [35]. Клиренс ремнантов ЛОНП (основной структурный белок апоВ100) из кровотока осуществляется через ЛНП-рецепторы. После связывания с рецепторами ремнантные частицы подвергаются дальнейшей липолитической трансформации, либо интернализируются гепатоцитами с последующей деградацией в лизосомах [8][36].
ХМ и ЛОНП в процессе модификации (уменьшения в размерах и постепенного обогащения ХС при истощении запасов ТГ) превращаются в атерогенные ЛП частицы. Длительная циркуляция ремнантов ТГ-богатых ЛП в кровотоке приводит к их большему обогащению ХС. Установлено, что ремнантные частицы могут переносить в четыре раза больше ХС (до ~8600 молекул) на одну частицу, чем одна частица ЛНП (2000-2700 молекул ХС), поэтому они классифицируются относительно ЛНП как более атерогенные частицы [8][31][37].
Следует подчеркнуть, что в качестве атерогенного фактора рассматриваются ремнантные частицы, обедненные ТГ, но обогащенные ХС в процессе модификации, с диаметром <70 нм (сюда не входят вновь секретируемые, но нелиполизированные ХМ и очень крупные ЛОНП) [17]. В случае неэффективного метаболизма подобных ремнантов активизируется их нерецепторный захват скэвенджер рецепторами на макрофагах, который является нерегулируемым (идет прямой захват макрофагами ремнантных частиц без их предварительной окислительной модификации, в отличие от ЛНП, которые, напротив, подвергаются модификации перед их захватом макрофагами).
Ремнантные частицы, содержащиеся в плазме крови, на поверхности эндотелия артерий взаимодействуют с гепарин сульфат — протеогликанами (взаимодействие усиливается за счет присутствия на поверхности частиц апоCIII и апоE), далее ремнанты мигрируют через эндотелий (путем активного трансцитоза) в субэндотелиальное пространство и там задерживаются длительное время, вызывая эффекты, свойственные окисленным ЛНП, а также сами подвергаются окислительной модификации, приобретая цитотоксические свойства [38][39]. В конечном итоге ремнантные частицы поглощаться макрофагами и гладкомышечными клетками, превращая их в "метаболически активные клетки" с трансформацией в пенистые клетки (рассматриваются в качестве маркера атеросклеротического поражения артерий) [40].
Задержка ремнантов ТГ-богатых ЛП в артериях вызывает выраженные дезадаптивные реакции, которые лежат в основе формирования и прогрессирования атеросклеротических бляшек. В атероме человека были идентифицированы ЛП частицы, содержащие как апоВ48, так и апоВ100 [41][42]. Показано, что примерно 50% ХС, содержащегося в атеросклеротической бляшке у человека, поступает из ремнантов ТГ-богатых ЛП, что подтверждает вовлечение этих частиц в атерогенез [42-44]. При ГТГ (повышенном уровне ТГ-богатых ЛП/ремнантов) изменяется количественный и качественный состав других ЛП в сторону проатерогенной направленности [8][45]:
— увеличивается образование мелких и плотных частиц ЛНП (в т.ч. при нормальном или низком уровне ХС ЛНП) с потенциально повышенной атерогенностью;
— снижается уровень ХС ЛВП и усиливается формирование дефектных мелких, плотных частиц ЛВП с нарушенным липидомом и протеомом, что ослабляет атеропротективное действие ЛВП, включая ухудшение обратного транспорта ХС, подавление антиоксидантной и противовоспалительной активности.
Генетические исследования подтверждают роль, например, апоCIII и ANGPTL-3 в развитии атеросклероза. Это способствует пониманию механизмов атерогенеза на основе ГТГ и позволяет определить новые терапевтические мишени. Показано, что апоCIII, ассоциированный с ТГ-богатыми ЛП, активирует инфламмасому NLRP3 в моноцитах человека; это может препятствовать регенерации эндотелия in vivo.
Атерогенный и тромбогенный механизмы ТГ-богатых ЛП довольно сложны. Среди атеротромбогенных свойств ремнантов ТГ-богатых ЛП, подтвержденных в исследованиях, можно выделить [8][17][46]:
— повышение проницаемости эндотелия артерий, активация апоптоза эндотелиальных клеток и подавление выработки окиси азота с развитием дисфункции эндотелия;
— усиление эндотелиальной экспрессии внутриклеточных адгезивных молекул-1 (ICAM-1) и сосудистых адгезивных молекук-1 (VСAM-1);
— увеличение продукции активных форм кислорода;
— повышение вязкости крови, уровней тромбоксана А2 — активатора тромбоцитов, фибриногена, активности VII и XII факторов коагуляции, секреции и активности ингибитора активатора плазминогена-1 — ключевого регулятора образования тромбов в эндотелиальных клетках;
— выраженную провоспалительную активность с повышенной секрецией проапоптотических цитокинов, фактора некроза опухоли α и интерлейкина 1-β.
В наблюдательном исследовании из Копенгагена (n=60608) было установлено, что каждый 1 ммоль/л повышения уровня ХС ремнантов в плазме крови ассоциировался с возрастанием концентрации высокочувствительного С-реактивного белка (вчСРБ) на 37%, а каждый 1 ммоль/л увеличения ХС ЛНП — с повышением вчСРБ только на 7% [47]. При применении в данном исследовании метода менделевской рандомизации результаты оказались иными: содержание вчСРБ увеличивалось на 28% на каждый 1 ммоль/л роста уровня ХС ремнантов в плазме крови, тогда как связи между маркером воспаления и ХС ЛНП обнаружено не было. Известно, что воспаление — важный компонент развития атеромы, ее надрыва и разрыва. Связь ГТГ с процессами воспаления, в т.ч. в стенке сосуда, становится очевидной и в определенной степени может объяснить сохранение остаточного ССР у пациента при нормализации уровня ХС ЛНП.
В исследовании PESA (Progression of Early Subclinical Atherosclerosis; 3754 лиц с низким и умеренным ССР, средний возраст 45,5 лет) сообщалось о связи ГТГ (уровень ТГ ≥1,7 ммоль/л) с субклиническим некоронарным атеросклерозом (отношение шансов (ОШ) 1,35; 95% доверительный интервал (ДИ): 1,08-1,68; р=0,008) [48]. Атеросклеротические бляшки выявлялись у 58% лиц с ГТГ при низком и умеренном ССР. Связь ГТТ с наличием субклинического атеросклероза подтвердилась как у лиц с повышенным уровнем ХС ЛНП в плазме крови (ОШ 1,42; 95% ДИ: 1,11-1,80; p=0,005), так и с нормальным его содержанием (ОШ 1,85; 1,08-3,18; p=0,008). На каждый 0,11 ммоль/л повышения уровня ТГ в плазме крови у лиц с целевым значением ХС ЛНП вероятность выявления атеромы увеличивалась на 4% (р=0,041).
В другом исследовании у пациентов с оптимальным уровнем ХС ЛНП (<1,8 ммоль/л) на каждый 1 ммоль/л повышения концентрации ХС ремнантов ТГ-богатых ЛП вероятность выявления коронарного атеросклероза (по данным компьютерной томографической коронарной ангиографии) возрастала в 3,87 раза (95% ДИ: 1,34-7,55; p=0,004) [49]. В исследовании Бубновой М. Г. и др. была обнаружена статистически значимая прямая корреляционная связь между уровнем ТГ-богатых ЛП/ремнантов (выделяемых методом ультрацентрифугирования) и степенью поражения коронарных артерий атеросклерозом (индексом стенозов и числом пораженных артерий по данным коронароангиографии) [50]. При этом наиболее высокие уровни ТГ-богатых ЛП/ремнантов встречались у лиц с тяжелым атеросклеротическим поражением артерий сердца. Elshazly MB, et al. по результатам метаанализа (5754 пациентов из 10 интервенционных исследований) продемонстрировали связь выраженного прогрессирования атеросклероза в коронарных артериях в большей степени с повышенным (>0,65-0,78 ммоль/л) уровнем ХС ремнантов, чем с концентрацией ХС ЛНП или апоВ в течение 24 мес. наблюдения [51]. Вовлечение ремнантов ТГ-богатых ЛП в прогрессирование атеросклероза (по данным коронароангиографии) подтверждается рядом проспективных исследований (с наблюдением от 2 до 5 лет) как у мужчин, так и у женщин [52-54].
Итак, повышенную концентрацию ТГ (ТГ-богатых ЛП/ремнантов) в плазме крови можно рассматривать как фактор "остаточного атеросклеротического риска". Частично объяснить этот факт можно доказанной заметной связью ГТГ с сосудистым воспалением. Очевидно, что уровень ТГ (ТГ-богатых ЛП/ремнантов) является значимым в идентификации пациентов, особенно с многососудистым поражением артерий, т.е. может выступать в роли маркера ангиографически верифицированного коронарного атеросклероза и ИБС.
ХС ЛНП — это первичная мишень для профилактики АССЗ. Действительно у большинства людей количество частиц ЛНП в плазме крови в ~3-10 раз больше, чем количество ТГ-богатых ЛП, также ЛНП более длительно циркулируют в плазме крови (в среднем 2,5-3,5 дня), чем ХМ и ЛПОНП (4-13 часов для лиц с ГТГ). Натощак во фракции ЛОНП + ЛПП находится не менее 30% ХС от его общего количества, определяемого во всей фракции апоВ-содержащих ЛП. Очевидно, что атерогенную опасность ТГ-богатых ЛП нельзя приравнивать к их меньшему количеству в плазме крови. При сравнении атерогенных потенциалов ремнантов ТГ-богатых ЛП и частиц ЛНП необходимо учитывать ряд факторов: время пребывания ЛП в кровотоке, загруженность их молекулами ХС, скорость проникновения и удержания в интиме артериальной стенки, восприимчивость к модификации in situ, скорость поглощения макрофагами и склонность к образованию пенистых клеток [8, 55]. Даже при неопределенности относительно точной доли ХС в ремнантных частицах (отчасти из-за методологических проблем [16]) длительное поддержание повышенного уровня ТГ в постпрандиальный период (≥8 ч) приводит к продолжительному и интенсивному воздействию ремнантных частиц на стенку артерий [56][57].
Ключевые положения
За последние три десятилетия были накоплены эпидемиологические и генетические данные, подтверждающие концепцию причинно-следственной связи ГТГ (ТГ-богатых ЛП/ремнантов) с повышенным риском развития АССЗ, включая ИМ, инсульт, стеноз аортального клапана, а также с сердечно-сосудистой и общей смертностью [7][58-67]. Крупные метаанализы показали, что повышенный уровень ТГ — независимый фактор развития АССЗ, риск которых увеличивается от 1,57 (95% ДИ: 1,10-2,24) до 1,8 (1,49-2,19) [61][68][69].
Ретроспективный продольный анализ результатов итальянского исследования (n=158042) выявил при высоком уровне ТГ (1,7-5,6 ммоль/л) повышение относительного риска (RR) развития всех АССЗ в 1,61 раза (95% ДИ: 1,43-1,82; р<0,001) и общей смертности в 1,49 раз (1,36-1,63; р<0,001), а при очень высоком уровне ТГ (>5,6 ммоль/л) — в 2,3 раза (1,02-5,18; р<0,05) и 3,08 раза (1,46-6,50; р<0,01), соответственно (относительно нормального уровня ТГ <1,7 ммоль/л) [70]. Данные когортного исследования с включением >1,5 млн чел. подтвердили небольшое, но значимое увеличение (на 10-20%) риска развития ИМ при умеренной и тяжелой ГТГ (уровень ТГ в пределах от 1,7 до 10,0 ммоль/л) после коррекции по другим факторам риска [71].
Данные 8,8-летнего наблюдения, полученные на когорте Корейской национальной службы медицинского страхования (n=15672028), продемонстрировали логарифмическую линейную связь между уровнем ТГ и сердечно-сосудистой смертностью [67]. Эта связь прослеживалась до уровня ТГ 0,56 ммоль/л и сохранялась после поправки на традиционные факторы риска развития АССЗ (индекс массы тела, артериальное давление, СД, уровни ХС ЛНП и ХС ЛВП, пол, возраст). Каждое двукратное повышение концентрации ТГ в плазме крови ассоциировалось с ростом RR всех ССЗ (на 10%), ИБС (на 22%), острого ИМ (на 24%) и ишемического инсульта (на 10%). Повышение RR смерти от АССЗ при ГТГ сохранялось и в когорте лиц (n=5480875) с низким уровнем ХС ЛНП (<2,6 ммоль/л). У лиц моложе 45 лет при двукратном росте уровня ТГ в плазме крови RR смертности от ИБС и ИМ были значимо выше, чем у лиц 65 лет и старше.
В рамках крупного когортного исследования (5688055 лиц) с наблюдением 7,1 лет также сообщалось о значимости ГТГ для развития сердечно-сосудистых осложнений (ССО) в молодом возрасте (20-39 лет): RR смертности возрастал в 1,79 раза (95% ДИ: 1,71-1,87), ИМ — в 2,48 раз (2,33-2,64) и инсульта — в 2,53 раза (2,34-2,73) [72]. Именно ГТГ определялась в качестве единственного независимого предиктора развития всех ССО (RR 1,20; 95% ДИ: 1,14-1,26; p<0,001). Результаты выше представленных и других исследований демонстрируют, что для поддержания более низкого ССР в течение жизни желательно сохранять уровень ТГ в пределах 1,12 ммоль/л, в т.ч. у молодых [73-75].
ГТГ, определяемая в образцах крови после еды (т.е. постпрандиальная), рассматривается в качестве маркера повышенного содержания ХС ремнантов ХМ/ЛОНП. В исследовании Copenhagen City Heart Study (13981 пациентов, не принимавших гиполипидемическую терапию; наблюдение 27 лет) указывается на связь постпрандиальной ГТГ с высоким риском развития смертности, ИМ и ИБС как у мужчин, так и у женщин [78]. Анализ объединенных исследований Copenhagen City Heart Study и Copenhagen General Population Study (~100 тыс. человек) показал возрастание RR развития ССО при уровне ТГ не натощак >6,6 ммоль/л (против уровня ТГ не натощак <0,8 ммоль): ИМ — в 5,1 раза, ИБС — в 3,2 раза, ишемического инсульта — в 3,3 раза и смерти от всех причин — в 2,2 раза [63]. В другом исследовании повышение постпрандиального уровня ТГ >5 ммоль/л (vs уровня ТГ не натощак <1 ммоль/л) ассоциировалось с увеличением RR развития ИМ в 4,6 раза у мужчин и в 16,8 раза у женщин, а ишемического инсульта — в 3,2 раза и 5,1 раза, соответственно [76][77].
В проспективном наблюдательном когортном исследовании Women's Health Study (n=26509) у здоровых женщин с ГТГ, определяемой в период от 2 до 4 ч после приема пищи, RR развития ССО увеличивался в 4,48 раза (независимо от традиционных факторов риска, других ЛП и маркеров инсулинорезистентности) [78]. Доказанная независимая связь ТГ (ТГ-богатых ЛП) с развитием ССО (ИМ, инсульта, сердечно-сосудистой смертности), с одной стороны, и достоверная их взаимосвязь с тромбогенным потенциалом крови, с другой стороны, позволяет рассматривать ГТГ как мощный липидный фактор тромбогенеза.
По данным двух исследований (n=113554) при высокой (≥5 ммоль/л) постпрандиальной ГТГ (относительно уровня ТГ не натощак <1 ммоль/л) возрастает вероятность развития (RR 2,59; 95% ДИ: 1,48-4,54) сердечной недостаточности (СН) [79]. В 5-летнем наблюдении, включавшем 46362 пациентов с СД и/или АССЗ, лечившихся статинами, риск появления новых случаев СН увеличивался на 19% (95% ДИ: 1,134-1,252; р<0,001) при уровне ТГ натощак ≥1,7 ммоль/л и на 24% (1,160-1,315; р<0,001) при уровне ТГ 2,3-5,6 ммоль/л (сравнение с уровнем ТГ <1,7 ммоль/л) [80].
Значение ХС ремнантов. В ряде исследований была продемонстрирована связь повышенного уровня ХС ремнантов (как расчетного, так и прямо измеряемого в плазме крови) с развитием АССЗ, включая ИМ, ишемический инсульт, ИБС, периферический атеросклероз и общую смертность, причем этот характер связи не зависел от концентрации ХС ЛНП в плазме крови [81-88].
В исследовании Copenhagen General Population (n=103221; наблюдение 9,5 лет) при высоком (>0,8 ммоль/л) уровне ХС ремнантов (против низкого <0,5 ммоль/л) RR развития ИМ повышался в 1,6 раза (95% ДИ: 1,5-1,8), АССЗ — в 1,4 раза (1,3-1,5) и общей смертности — в 1,1 раза (1,0-1,1) [86]. При сочетании высокого уровня ХС ремнантов с повышенной концентрацией вчСРБ (>1,5 мг/л) риски ССО существенно возрастали: ИМ — в 2,2 раза (95% ДИ: 1,9-2,7), АССЗ — в 1,9 раза (1,7-2,2) и общей смертности — в 1,4 раза (1,3-1,5). Wadström BN, et al. изучали влияние ХС ремнантов (определяемого расчетным методом) на риски развития АССЗ у 106937 пациентов из исследования Copenhagen General Population Study (CGPS; наблюдение 9 лет) и 13974 пациентов из исследования Copenhagen City Heart Study (CCHS: наблюдение 24 года) [89]. Повышение уровня ХС ремнантов в плазме крови ≥1,5 ммоль/л (vs <0,5 ммоль/л) сопровождалось увеличением RR развития ИМ в 4,2 раза (95% ДИ: 2,9-6,1) в исследовании CGPS и в 2,6 раза (1,8-3,8) в исследовании CCHS; ишемического инсульта — в 1,8 (1,4-2,5) и 2,1 (1,5-3,1) раза, соответственно; но более значимо возрастал RR развития периферической артериальной болезни — в 4,8 (3,1-7,5) и 4,9 (2,9-8,5) раз, соответственно.
По данным метаанализа, включавшего 31 исследование, при повышенном уровне ХС ремнантов в плазме крови (против его низкого значения) RR развития ССО увеличивался на 53% (95% ДИ: 1,41-1,66; p<0,0001), ИБС — на 41% (1,19-1,67; p<0,0001), инсульта — на 43% (1,24-1,66; p<0,0001), сердечно-сосудистой смертности — на 83% (1,53-2,19; p<0,0001) и общей смертности — на 39% (1,27-1,50; p<0,0001) [90]. Заметное увеличение риска ССО имелось в группе моложе 65 лет (RR 1,39; 95% ДИ: 1,14-1,70, p<0,0001 vs ≥65 лет RR 1,20; 1,16-1,25, p<0,0001). На каждый 1 ммоль/л повышения уровня ХС ремнантов RR развития ССО увеличивался на 15% (95% ДИ: 1,08-1,22) и ИБС — на 58% (1,12-2,23).
У пациентов, госпитализированных с острым коронарным синдромом (ОКС), повышенный (≥0,78 ммоль/л) уровень ХС ремнантов в плазме крови выявлялся у 45,9% (из n=7479) [91]. После выписки из стационара (медиана наблюдения 57 мес.) повышение его значения до 1,55 ммоль/л и выше приводило к увеличению RR сердечно-сосудистой смертности на 49% (95% ДИ: 1,08-2,06; р=0,016) и повторной госпитализация из-за СН на 55% (95% ДИ: 1,14-2,11; р=0,005).
В ряде исследований подтверждается позиция, что повышение RR развития смерти от всех причин в большей степени обусловлено ростом именно концентрации ХС ремнантов в плазме крови, а не уровней ХС ЛНП [83][92]. Известно, что повышенный уровень общего ХС связан с ростом смертности от ССЗ, но не от рака или других некоронарогенных причин, тогда как ГТГ может значимо повышать как сердечно-сосудистую смертность, так и смертность от других некоронарогенных причин.
В рамках исследования Copenhagen General Population (n=87192; возраст 20-69 лет; наблюдение 13 лет) была выявлена связь повышенного (≥1,0 ммоль/л) уровня ХС ремнантов (сравнение со значением <0,5 ммоль/л) с увеличением в 2,2 раза (95% ДИ: 1,3-3,5) частоты сердечно-сосудистой смертности; в 4,4 раза (1,6-11) смертности от ИБС; в 2,1 раза (1,4-3,3) всех случаев смерти от некоронарогенных причин, включая возрастание в 8,4 раза (2,0-34) смертности от инфекционных заболеваний и в 9,1 раза (1,9-43) смертности от эндокринологических заболеваний [93].
Доказательства значимости ГТГ для развития АССЗ в генетических исследованиях. Причинно-следственная связь ТГ (точнее ТГ-богатых ЛП и их ремнантов) с риском развития АССЗ подтверждается исследованиями с менделевской рандомизацией. Так, у лиц с генетически детерминированным высоким уровнем ТГ-богатых ЛП/ремнантов определяется высокий RR (от 1,14 до 4,0 в зависимости от пола, расы после поправки на другие факторы, в т.ч. на другие ЛП) развития CCЗ, включая ИБС, ИМ и смертность [76][81][94-96].
Многофакторный анализ индивидуальных данных (генетических и стандартных липидных показателей, включая апоВ) участников из Биобанка Великобритании (n=502460) показал, что уровни ТГ-богатых ЛП/ремнантов тесно и независимо связаны с риском развития ИБС в моделях, скорректированных по апоB. Вероятность развития ИБС на каждый 1 ммоль/л повышения ТГ-богатых ЛП/ремнантов оказалась выше (ОШ 2,59; 95% ДИ: 1,99-3,39), чем при повышении ХС ЛНП на эту же величину (ОШ 1,37; 95% ДИ: 1,27-1,48) [97].
Исследования менделевской рандомизации, сосредоточенные на вариантах генов, кодирующих белки — АРОAV, APOCIIII, LPL и ангиопоэтин-подобные белки 3, 4 и 8 (ANGPTL3, ANGPTL4 и ANGPTL8), представили высокоинформативные доказательства связи ТГ-богатых ЛП и их ремнантов с риском развития АССЗ [98-104]. Определено, что при генетически обусловленном повышении уровня ХС ремнантов в плазме крови на 1 ммоль/л RR развития ИБС возрастает достоверно выше (в 2,8 раза), чем в наблюдательных исследованиях (только в 1,4 раза) [81]. Для вариантов гена APOA5 удвоение генетически повышенного уровня ХС ремнантов увеличивает риск ИМ в 2,2 раза vs 1,7 раз в наблюдательных исследованиях, а удвоение генетически обусловленного повышения уровня ТГ — в 1,9 раза и 1,6 раза, соответственно [94]. У лиц с мутаций гена LPL риск смерти от всех причин при высоком уровне ТГ возрастает двукратно, а в наблюдательном исследовании — только в 1,2 раза [105]. У гетерозигот с редкими вариантами гена APOC3, приводящими к потере функции, на фоне снижения (в среднем на 39-44%) уровня ТГ в крови происходит заметное снижение (на 40-41%) риска развития АССЗ [99][106].
Перечисленные выше факты служат дополнительным доказательством причинно-следственной связи ТГ (ТГ-богатых ЛП и их ремнантов) с риском развития АССЗ на протяжении всей жизни.
Остаточный риск АССЗ, обусловленный ТГ (ТГ-богатыми ЛП). Концепция так называемого "остаточного ССР", связанная с липидами и ЛП, основывается на развитии у пациента серьезных ССО даже при оптимальном контроле уровня ХС ЛНП [107]. В значительной степени сохранение остаточного риска развития ССЗ, обусловленных атеросклерозом, объясняют повышенным содержанием в плазме крови ТГ (ТГ-богатых ЛП и их ремнантов) и/или низким уровнем ХС ЛВП.
В исследовании NHANES (National Health and Nutrition Examination Survey) при анализе результатов было установлено, что у пациентов с уровнем TГ ≥1,69 ммоль/л, несмотря на прием статинов, RR развития основных ССО был выше 26% (95% ДИ: 1,19-1,34) относительно пациентов с уровнем ТГ <1,7 ммоль/л [108]. У пациентов с СД и уровнем ТГ 2,26-5,64 ммоль/л частота развития ССО оставалась высокой даже при нормализации уровня ХС ЛНП: риск нефатального ИМ повышался на 30% (95% ДИ: 1,08-1,58; р=0,006), инсульта — на 23% (1,01-1,49; р=0,037) и коронарной реваскуляризации — на 21% (1,02-1,43; р=0,027).
Достаточное количество исследований (большинство это post hoc анализы) демонстрируют взаимосвязь между уровнем ТГ и риском развития АССЗ/ССО у пациентов на гиполипидемической терапии [109-112]. В то же время снижение концентрации ТГ в плазме после лекарственной коррекции уровня ХС ЛНП ведет к дополнительному уменьшению риска ССО.
В исследовании PROVE IT-TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction 22) наблюдались пациенты с ОКС (n=4162), которые получали статины. У пациентов с низким (<1,7 ммоль/л) уровнем ТГ при сравнении с группой пациентов с ГТГ (ТГ ≥1,7 ммоль/л) риск развития повторных случаев ИБС снижался на 27% (95% ДИ: 0,62-0,87; р<0,001) в однофакторном анализе и на 20% (0,66-0,97; р=0,025) в скорректированном анализе [112]. Снижение концентрации ТГ на каждые 0,11 ммоль/л (10 мг/дл) приводило к уменьшению показателей смертности, заболеваемости ИМ или повторным ОКС на 1,6% (p<0,001) или на 1,4% (р<0,01) после поправки на ХС ЛНП или ХС не-ЛВП. Значимое снижение риска развития ССО на 41% (95% ДИ: 0,41-0,83; p=0,002) наблюдалось при достижении оптимального уровня трех параметров: ТГ (<1,7 ммоль/л), ХС ЛНП (<1,8 ммоль/л) и вчСРБ (<2,0 мг/л).
Результаты исследования IDEAL (Incremental Decrease in End Points Through Aggressive Lipid Lowering) и TNT (Treating to New Targets) продемонстрировали сохранение высокого риска развития ССО при наличии ГТГ (после поправки на ХС ЛВП и величины отношения апоВ/апоАI) у пациентов с ИБС, получавших статины [113]. Влияние ГТГ на краткосрочный и долгосрочный прогнозы пациентов, перенесших ОКС и получавших гиполипидемическую терапию, оценивалось в исследованиях MIRACL (Myocardial Ischemia Reduction with Acute Cholesterol Lowering, n=1501; наблюдение 16 нед.) и dal-OUTCOMES (A Study of RO4607381 in Stable Coronary Heart Disease Patients With Recent Acute Coronary Syndrome, n=15817; наблюдение 31 мес.), соответственно [110]. В исследовании MIRACL при переходе от уровня ТГ от ≤1,5 ммоль/л к уровню ТГ >2,2 ммоль/л RR развития ССО увеличивался на 50% (95% ДИ: 1,05-2,15; р=0,03) в течение 16 нед. наблюдения. В исследовании dal-OUTCOMES при переходе от значения ТГ ≤0,9 ммоль/л к уровню ТГ >2,0 ммоль/л (175 мг/дл) RR развития ССО повышался на 61% (95% ДИ: 1,34-1,94; p<0,001) в течение 31 мес. наблюдения. В обоих исследования связь ГТГ с риском ССО не зависела от концентрации ХС ЛНП.
Статины способны ускорять выведение ремнантов ЛОНП и ЛНП из кровотока и, следовательно, снижать частоту развития ССО при ГТГ, но, по-видимому, этого недостаточно. Для снижения остаточного ССР необходимо воздействовать на концентрацию ТГ и снижать ее до рекомендуемого целевого значения. Это важно, поскольку уровень ТГ независимо предсказывает краткосрочный и долгосрочный риски развития АССЗ у пациентов как в первичной, так и во вторичной профилактике, в т.ч. у принимающих гиполипидемические препараты. Следовательно, повышенная концентрация ТГ в плазме крови представляет собой потенциальную цель для лечения.
Ключевые положения
С целью нивелирования повышенного остаточного ССР пациента важно определить терапевтические подходы для снижения концентрации ТГ в плазме крови. Чтобы ускорить получение позитивного результата предлагается придерживаться определенной схемы обследования и маршрутизации пациента с ГТГ (рис. 2). Исходно проводится обследование пациента разными специалистами для выявления состояний, которые могут быть связаны с ГТГ: АССЗ, метаболического синдрома, ожирения, СД 2 типа, острого/хронического панкреатита в анамнезе, ХБП, неалкогольной жировой болезни печени и др. У пациентов исключаются вторичные причины ГТГ, включая оценку сопутствующей терапии, способной повышать уровень ТГ в плазме крови. При осмотре пациента обращается внимание на наличие ксантом, ксантелазм, твердого ретинального экссудата и других признаков ГТГ при осмотре глазного дна, гепатоспленомегалию. Осматриваются ладони рук для обнаружения ладонных полосок; локти, колени и ягодицы для выявления эруптивных ксантом; тыльные стороны ладоней и ступней, претибиальные бугорки и ахилловы сухожилия.
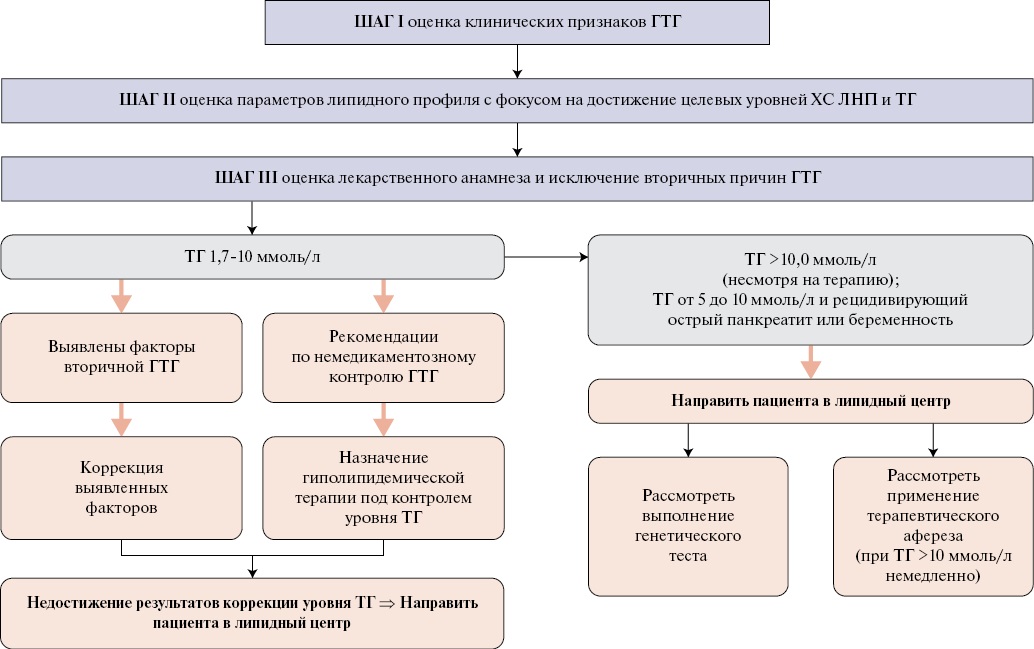
Рис. 2. Поэтапное обследование и маршрутизация пациента с ГТГ.
Сокращения: ЛНП — липопротеины низкой плотности, ГТГ — гипертриглицеридемия, ТГ — триглицериды, ХС — холестерин.
Необходимо подтвердить наличие ГТГ при взятии образцов крови натощак (после ночи и более чем 10-часового голодания). Для диагностики I, III и V типов ГЛП полезно исследовать плазму или сыворотку крови, оставленную на 24 ч при 4 °С в холодильнике (появление сливообразного слоя наблюдается при I типе ГЛП), провести электрофорез ЛП (при выполнении этого метода в лаборатории для исключения ГЛП III типа) и фенотипирование апоЕ.
Пациентов с уровнем ТГ >10,0 ммоль/л или с уровнем ТГ от 5 до 10 ммоль/л при рецидивирующем остром панкреатите или беременных женщин рекомендуется направлять в липидный центр для проведения генетического анализа и терапевтического афереза. Недостижение результатов коррекции повышенного (до 5,0 ммоль/л) уровня ТГ предложенными методами также является основанием для направления пациента в липидный центр. При этом важно сохранить взаимодействие между лечащим врачом, направившим пациента в липидный центр, и врачом липидного центра.
Изменение образа жизни (коррекция питания, отказ от курения, регулярная физическая активность, ограниченное потребление алкоголя и поддержание здорового веса) — это важнейший компонент снижения риска ССЗ при ГТГ, в т.ч. совместно с применением гиполипидемических препаратов. Модификация образа жизни должна быть максимально персонализирована и направлена на основные триггеры, которые вносят наибольший вклад в ГТГ.
Первый шаг в ведении пациентов с ГТГ — это предоставление консультации по образу жизни, основанной на фактических данных. Например, у пациентов с СД следует обратить внимание на контроль гликемии, поскольку это может положительно влиять на поддержание концентрации ТГ в норме. Для пациентов с избыточной массой тела или ожирением редукция веса считается наиболее эффективным изменением образа жизни с целью снижения уровня ТГ. Уменьшение массы тела на 1 кг сопровождается снижением уровня ТГ в среднем на 2% [114]. Снижение массы тела на 5-10% приводит к снижению концентрации ТГ в плазме крови на 20% (у некоторых пациентов до 50-70%) [14].
Оптимизация рациона питания в сочетании с регулярной аэробной физической активностью может привести к снижению уровня ТГ на 20-50%. Регулярные аэробные тренировки умеренной интенсивности способны снижать уровень ТГ натощак в плазме крови до 11%, а постпрандиальный уровень ТГ — от 21 до 27% [115-117]. В исследовании Аронова Д. М. и др. показана возможность длительной (до ≥40 мин) динамической физической нагрузки умеренной интенсивности увеличивать клиренс ТГ-богатых ЛП частиц у здоровых лиц на 84,5% и больных ИБС на 48,1% [117].
Для пациентов с ГТГ (уровнем ТГ натощак ≥1,7 ммоль/л и <5,0 ммоль/л) высокого и очень высокого ССР в первичной и вторичной профилактике выбор первоначальной гиполипидемической терапии основывается на необходимости достижения целевого уровня ХС ЛНП в соответствии с величиной ССР.
При тяжёлой ГТГ (уровень ТГ ≥5,0 ммоль/л и особенно при ТГ >10 ммоль/л) следует проводить терапию, направленную на снижение избытка как ХМ, так и ЛОНП. Для таких пациентов требуется разработать индивидуальную программу, включающую более строгие ограничения в питании (совместное ведение пациента с диетологом) и изменения образа жизни с назначением лекарственных средств.
Пациентам с уровнем ТГ >10 ммоль/л рекомендуется очень строгий подход и значительные ограничения в питании. Известно, например, что диеты с высоким содержанием сахара или насыщенных жиров увеличивают синтез апоCIII (подавляющего активность периферической ЛПЛ) [31]. Если насыщенные ЖК стимулируют экспрессию апоCIII, то полиненасыщенные ЖК (ПНЖК), напротив, ее подавляют. Также у пациентов с семейной хиломикронемии (ГЛП I типа) ограничена эффективность лекарственной терапии в снижении экстремального уровня ТГ в плазме крови.
Итак, большинству пациентов следует стремиться к целевому уровню ТГ <1,7 ммоль/л, пациентам с тяжелой ГТГ к достижению уровня ТГ <5,6 ммоль/л (как более реалистичного). Стратегия ведения пациентов с ГТГ представлена на рисунке 3.
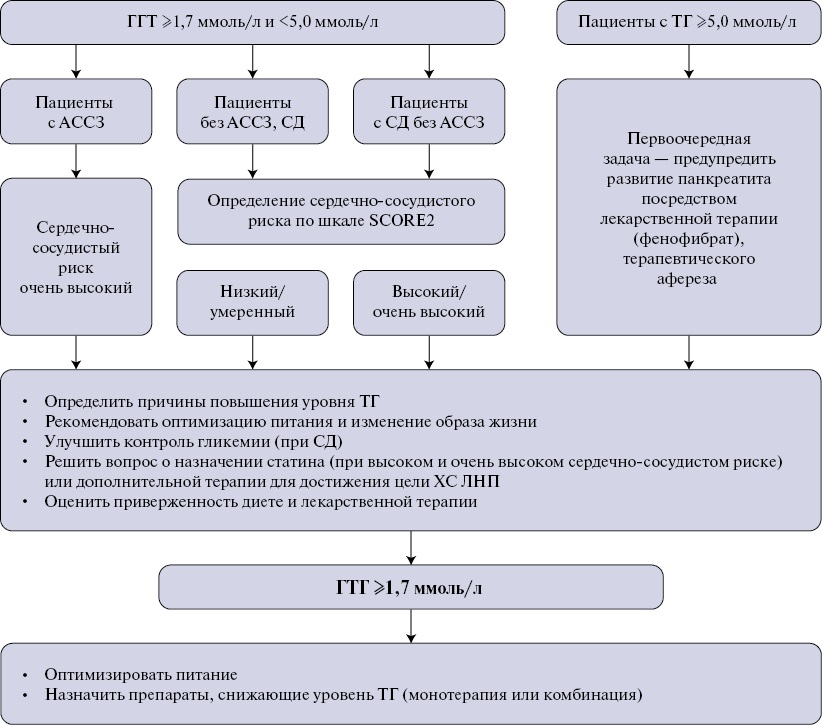
Рис. 3. Алгоритм ведения пациентов с ГТГ (согласованная позиция экспертов).
Сокращения: АССЗ — атеросклеротические сердечно-сосудистые заболевания, ГТГ — гипертриглицеридемия, ЛНП — липопротеины низкой плотности, СД — сахарный диабет, ТГ — триглицериды, ХС — холестерин.
Для пациентов с уровнем ТГ от 5,0 ммоль/л до 10,0 ммоль/л доля общего жира в рационе должна составлять 20-25% от суточной калорийности, а для пациентов с уровнем ТГ ≥10 ммоль/л — 10-15% [14]. Рекомендуется равномерное распределение потребления жира между приемами пищи с целью избегания резкого подъема ТГ [13][118]. Необходимо, чтобы 2-4% от калорийности рациона приходилось на долю α-линоленовой и линолевой кислот, для обеспечения суточной потребности организма в незаменимых ЖК [14][118]. Пищевые источники α-линоленовой кислоты — это семена чиа и льна, льняное масло, грецкие орехи, масло грецкого ореха, соевые бобы, тофу, а источники линолевой кислоты — цельные злаки [119]. Пациентам с панкреатитом в анамнезе рекомендуется более строгое ограничение жиров в рационе — <5% от общей калорийности [14].
Снижение уровня ТГ в плазме крови было наибольшим при диете с максимальным ограничением доли углеводов (<30-40% от суточной калорийности рациона) в сочетании с потерей массы тела [120]. Сравнительный анализ влияния низкоуглеводной диеты и низкожирового рациона в краткосрочном периоде (8-нед.) продемонстрировал снижение уровня ТГ на 18% только при низкоуглеводном типе питания (<20% от суточной калорийности рациона) [121]. Снижение концентрации ТГ в плазме крови наблюдается на диете с более высокой долей общего белка (>25-30% от суточной калорийности), чем на рационе с долей белка 15-18% (одновременно это сочетается с потерей массы тела) [122].
Пациентам с уровнем ТГ 5,6-10,0 ммоль/л рекомендуется потреблять <5% калорий в виде добавленного сахара, а пациентам с уровнем ТГ >10,0 ммоль/л следует отказаться от потребления добавленного сахара. Необходимо ограничить потребление фруктозы, главным источником которой является сахароза — дисахарид, содержащий глюкозу и фруктозу [123].
Высокое потребление фруктов (по сравнению с низким) лицами с ГТГ может приводить к снижению уровня ТГ до 21% (данные метаанализа) [124]. Предполагается, что увеличение потребления фруктов сопровождается и более высоким потреблением пищевых волокон, которые и обеспечивают сдерживающий эффект повышения ТГ. Употребление большого количества пищевых волокон снижает содержание ТГ в плазме крови. При этом пациентам с ГТГ рекомендуется потребление цельных несладких фруктов с высоким содержанием пищевых волокон (ягоды, яблоки) без добавленного сахара и отказ от потребления фруктовых соков. Для эффективного контроля ТГ, других липидов и ЛП плазмы крови рекомендуется потреблять от 25 до 40 г общего количества пищевых волокон, из них от 5 до 15 г растворимых волокон.
Потребление рыбы и морепродуктов рекомендуется не менее 2 раз в нед., в количестве не менее 280 г, что обеспечит поступление в организм примерно 450 мг эйкозапентаеновой кислоты (ЭПК; 20:5n-3) + докозагексаеновой кислоты (ДГК; 22:6n-3) в день. При этом лучше потреблять рыбу и морепродукты с минимальным содержанием контаминантов (ртути и диоксинов): треску, тилапию, креветки, сома, камбалу, пикшу, терпуга, сардины, лосось, скумбрию, сельдь2. Результаты системных обзоров и метаанализов рандомизированных клинических исследований (РКИ) убедительно демонстрируют липид-модифицирующий эффект потребления омега-3 ПНЖК со значительным снижением уровня ТГ у лиц с ГТГ (-0,39; 95% ДИ: -0,59 — -0,18; I²=17,2%) [125-127].
При хиломикронемии, в случае необходимости достижения требуемой энергоценности рациона и макронутриентного баланса, возможно применение специализированного продукта — масла среднецепочечных ТГ, который получается из кокосового и пальядрового масел путем гидролиза и последующего фракционирования. Вводить в рацион данный продукт следует постепенно, начиная с малых доз. Продукт содержит только ЖК средней цепи (каприновую и каприловую кислоты) в отличие от сырьевых продуктов кокосового и пальмоядрового масел, которые в своем составе одновременно имеют и длинноцепочечные ЖК [118].
Лицам с ГТГ следует ограничить потребление алкоголя до полного отказа. Коррекция рациона питания у лиц с ГТГ должна быть максимально индивидуализирована и учитывать исходный уровень ТГ. Предпочтение следует отдавать дифференцированному подходу к коррекции привычек питания у лиц с ГТГ. Суммированные рекомендации по модификации рациона у лиц с ГТГ представлены в Приложении 1.
Лекарственная терапия у пациентов высокого и очень высокого ССР с уровнем ТГ ≥1,7 ммоль/л и <5,0 ммоль/л начинается с оценки концентрации в крови ХС ЛНП и его соответствия целевому значению [1]. Для контроля уровня ХС ЛНП первоначально назначают статины в максимально переносимой дозе, а при недостижении цели переходят к комбинированной терапии посредством присоединения нестатиновых ХС-снижающих препаратов (эзетимиба, ингибиторов PCSK9).
Снижение уровня ТГ в плазме крови можно ожидать на любой дозе статина, однако каждое удвоение дозы статина дополнительно снижает уровень ТГ только от 2% до 4% [128]. Высокоинтенсивная терапия статинами может приводить к снижению уровня ТГ до 30% (табл. 3). Снижение уровня ТГ на фоне приема статинов происходит за счет уменьшения синтеза ЛОНП и стимуляции активности ЛНП-рецепторов на поверхности клеток, которые наряду с ХС ЛНП могут захватывать частицы, богатые ТГ. Более заметный гипотриглицеридемический эффект статинов проявляется при исходно повышенной концентрации ТГ в плазме крови.
Таблица 3
Липидные эффекты гиполипидемических препаратов [адаптировано из 1]
|
Код АТХ |
Препараты и суточные дозы |
Липидные эффекты |
|
C10AA Ингибиторы ГМГ-КоА-редуктазы |
Симвастатин (20-40 мг) Аторвастатин (10-80 мг) Розувастатин (5-40 мг) Питавастатин (1-4 мг) |
ХС ЛНП ↓ 20-60% ХС ЛВП ↑ 5-15% ТГ ↓ 7-30% ХС не-ЛВП ↓ 15-50% |
|
C10AX Другие гиполипидемические средства |
Эзетимиб (10 мг) |
ХС ЛНП ↓ 15-22% ХС ЛВП ↑ 1-2% TГ ↓ 5-10% ХС не-ЛВП ↓ 14-19% |
|
Эволокумаб (140 мг, подкожно, раз в 2 нед./420 мг, подкожно, 1 раз в мес.) Алирокумаб (75 мг, подкожно, раз в 2 нед.; 150 мг раз в 2 нед./300 мг раз в мес.) |
ХС ЛНП ↓ 50-70% ХС ЛВП ↑ 4-7% TГ ↓ 6-19% ХС не-ЛВП ↓ 20-50% |
|
|
C10AB Фибраты |
Фенофибрат (145 мг в сут.) |
ХС ЛНП ↓ 10-15% ХС ЛВП ↑ 10-20% TГ ↓ 20-50% ХС не-ЛВП ↓ 5-19% |
|
C10AX Другие гиполипидемические средства |
Омега-3 кислоты этиловых эфиров (2-4 г в сут.) |
ХС ЛНП ↓ 6% — ↑ 25% ХС ЛВП ↓ 5% — ↑ 7% TГ ↓ 20-45% ХС не-ЛВП ↓ 5-14% |
Сокращения: ЛВП — липопротеины высокой плотности, ЛНП — липопротеины низкой плотности, ТГ — триглицериды, ХС — холестерин.
Назначение препаратов (фибратов — производных фиброевой кислоты и этиловых эфиров омега-3 ПНЖК), целенаправленно снижающих уровень ТГ, рассматривают при повышении концентрации ТГ в плазме крови >2,3 ммоль/л. Выполненные клинические исследования подтверждают эффективность лекарственной терапии у пациентов с уровнем ТГ >2,3 ммоль/л [129]. При изолированном повышении уровня ТГ в диапазоне от 1,7 до 2,3 ммоль/л исходно требуется немедикаментозная коррекция, но возможно рассмотреть и назначение фенофибрата или омега-3 ПНЖК (рис. 4).
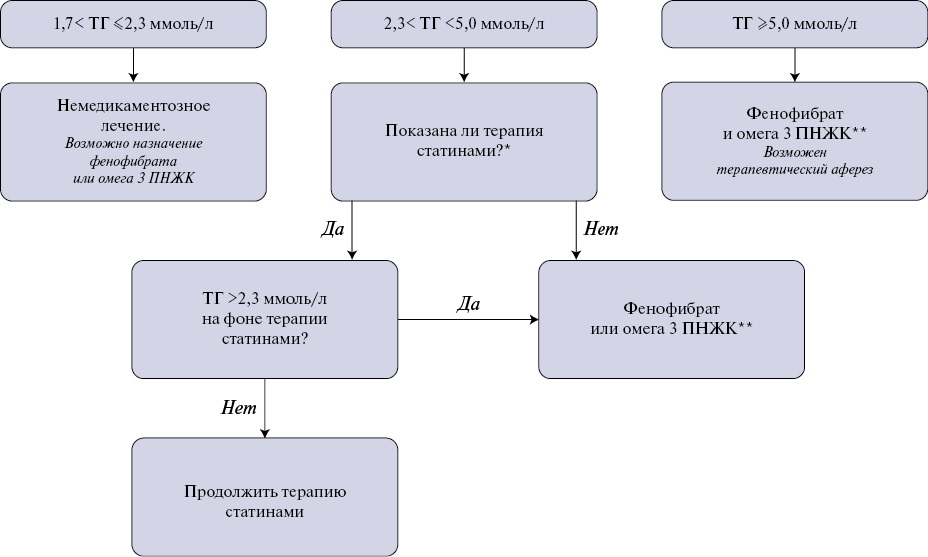
Рис. 4. Терапия ГТГ [адаптировано из 1].
Примечание: * — с учетом клинического фенотипа пациента, ** — омега 3 ПНЖК — омега 3 полиненасыщенных жирных кислот этиловые эфиры 90 в дозе 2-4 г/сут.
Сокращения: ПНЖК — полиненасыщенные жирные кислоты, ТГ — триглицериды.
Пациентам высокого и очень высокого риска, принимающим статины (или другие ХС-снижающие препараты) и достигающим уровень ТГ в пределах 1,7-2,3 ммоль/л, рекомендовано добавить препарат омега-3 ПНЖК в дозе до 2 г 2 раза/сут. [1]. Пациентам с уровнем ТГ >2,3 ммоль/л на терапии статином следует добавить фенофибрат (предпочтительно в одной таблетке, например, зарегистрированную комбинацию розувастатина с фенофибратом) или препарат омега-3 ПНЖК в дозе 2 г 2 раза/сут. Пациентам с уровнем ТГ >5,0 ммоль/л рекомендовано назначить фенофибрат и препарат омега-3 ПНЖК в дозе 2 г 2 раза/сут. При экстремальной ГТГ можно рекомендовать применение методов терапевтического афереза.
Многофакторность действия фибратов (в основном исследования проведены с фенофибратом) на ключевые компоненты системы обмена ЛП объясняется тем, что они являются агонистами ядерных рецепторов, активируемых пероксисомальным пролифератором (PPARα), расположенных в печени, мышцах, жировой ткани, сердце, почках, макрофагах и тромбоцитах [130][131]. Основная роль PPARα-рецепторов заключается в регуляции метаболизма липидов и ЛП, воспаления, функции эндотелия.
Фенофибрат после соединения с молекулой PPARα активирует её, образует активный комплекс (гетеродимер) с ретиноидным Х рецептором (RXR), действующий на экспрессию белков, регулирующих процессы обмена ЛП. При этом повышается экспрессия гена фермента ЛПЛ и генов ферментов, вовлеченных в β-окисление ЖК, снижается экспрессия гена апоСIII — ингибитора ЛПЛ и активируются гены основных белков ЛВП — апоАI и апоАII [130][131]. Противоатерогенные эффекты фенофибрата проявляются в снижении синтеза ТГ из ЖК, сборки и секреции ЛОНП, активации их липолиза при участии ЛПЛ. Этому также способствует снижение синтеза апоСIII — основного ингибитора ЛПЛ. Наряду с этим увеличивается синтез апоАI и апоАII — антиатерогенных белков ЛВП.
Под действием фенофибрата (микронизированная форма) уровень ХС ЛНП в плазме крови снижается умеренно (до 25%), тогда как содержание ТГ уменьшается значимо (до 50%) и концентрация ХС ЛВП увеличивается заметно (до 20%) [132-134]. Фенофибрат, повышая активность ЛПЛ и, следовательно, ускоряя катаболизм ТГ-богатых ЛП, обеспечивает сдвиг в размере и плотности ЛНП: от малых плотных наиболее атерогенных частиц ЛНП к субфракциям с более крупными частицами, имеющим хорошее сродство к ЛНП-рецепторам (количество таких частиц снижается до 51%). Этот эффект, направленный на коррекцию субфракционного спектра ЛНП, является специфической особенностью антиатерогенного действия фенофибрата и может рассматриваться как дополнительный механизм снижения риска АССЗ.
В Российском исследовании TRISTAN (152 клинических центра из 35 регионов) оценивалась гиполипидемическая эффективность и безопасность фенофибрата в дозе 145 мг у пациентов с метаболическим синдромом и ГТГ: 988 пациентов (46,4% женщины) с ИБС — 31,1%, ИМ в анамнезе — 10%, СД 2 типа — 26,8% [135]. Через 5-7 мес. после включения уровни ХС ЛНП в плазме крови снижались на 24,7%, ТГ — на 50,1%, ХС не-ЛВП — на 33,7%, а ХС ЛВП повышался на 23%. На протяжении всего исследования сохранялась высокая приверженность (97,8%) терапии фенофибратом.
В исследованиях показана способность фенофибрата подавлять воспалительный компонент атеросклеротического процесса, а именно снижать уровни вчСРБ (до 34%, а в исследовании TRISTAN до 39%), интерлейкина-6 (до 27%) и интерлейкина-1β (до 35%), фактора некроза опухоли-α (до 32%). Доказано, что фенофибрат уменьшает активность факторов, вовлеченных в процесс тромбообразования: фибриногена (до 21%), фактора коагуляции VII (до 19%) и ингибитора тканевого активатора плазминогена (до 26%). К положительным действиям фенофибрата следует отнести уменьшение в плазме крови концентрации мочевой кислоты (до 25%).
Позитивные липидные и плейотропные действия микронизированного фенофибрата реализуются в клинических эффектах. Одно из первых исследований этой категории — исследование DAIS (Diabetes Atherosclerosis Intervention Study), по результатам которого было доказано положительное влияние терапии фенофибратом на размер атеросклеротической бляшки [136]. Результаты РКИ FIELD (Fenofibrate Intervention and Event Lowering in Diabetes) и ACCORD (Action to Control Cardiovascular Risk in Diabetes) оказались еще более значимыми [137][139].
Первым крупнейшим РКИ по оценке клинической эффективности микронизированного фенофибрата у пациентов (n=9795) с СД 2 типа было исследование FIELD (наблюдение в среднем 5 лет) [137][139]. Достоверное снижение первичной конечной точки (нефатального ИМ и смерти от ИБС) на 27% (р=0,005) произошло только в группе пациентов с уровнем ТГ >2,3 ммоль/л и низким ХС ЛВП. В общей группе пациентов, получавших фенофибрат, при сравнении с группой плацебо наблюдалось снижение отношения рисков развития нефатального ИМ на 24% (p=0,01), всех ССО на 11% (р=0,035), стенокардии на 18% (р=0,04) и коронарной реваскуляризаций на 21% (p=0,035). Следует отметить, что исследуемым пациентам разрешалось применять статины, и к концу исследования наблюдалась диспропорция в количестве пациентов, которые их получили (17% в группе плацебо и 8% в группе фенофибрата; р<0,0001), что могло повлиять на частоту исходов в основной и контрольной группах.
Исследование NHIS-HEALS, выполненное в реальной клинической практике на когорте Корейской национальной службы медицинского страхования (10114 пациентов с СД 2 типа, наблюдение 3 года), продемонстрировало на терапии фенофибратом (против группы контроля) снижение отношения рисков первичной конечной точки (сердечно-сосудистой смертности, развития первого ИМ, инсульта или потребности в чрескожном коронарном вмешательстве) на 24% (р=0,01), инсульта на 38% (р=0,0015), сердечно-сосудистой смертности на 41% (р=0,0446) и смерти от всех причин на 56% [140].
В РКИ ACCORD у пациентов (n=5518) с СД 2 типа оценивалась эффективность одного из вариантов комбинированной терапии статина с фенофибратом (наблюдение 4,7 лет) в сравнении с монотерапией статином [138]. Добавление к терапии фенофибрата ассоциировалось со снижением частоты ССО (сердечно-сосудистой смертности, нефатального ИМ или инсульта) на 31% (р=0,032) в группе пациентов с атерогенной дислипидемией (уровнем ТГ >2,3 ммоль/л и низким ХС ЛВП <0,9 ммоль/л). При этом комбинированная терапия фенофибратом со статином (симвастатином) хорошо переносилась пациентами.
В когортном исследовании ECLIPSE-REAL (10705 пациентов с метаболическим синдромом; наблюдение 12 лет) 2156 участников получали комбинированное лечение (статин плюс фенофибрат) и 8549 участников — только статины [141]. Риск основных ССО был ниже на 26% (95% ДИ: 0,58-0,93; р=0,01) на комбинированной терапии по сравнению с монотерапией статинами. Значимость полученного результата сохранялась и при анализе в процессе лечения (RR 0,63; 95% ДИ: 0,44-0,92; р=0,02).
Неудачи исследования PROMINENT (Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts With diabeTes) с применением пемафибрата нельзя перекладывать на фенофибрат, поскольку эти лекарства по-разному модифицируют уровни липидов и ЛП плазмы крови: фенофибрат снижает уровни ХС ЛНП и апоВ, а пемафибрат, напротив, повышает их [142].
Показана способность фенофибратом предотвращать прогрессирование ретинопатии (исследования FIELD, ACCORD-EYE, Eclipse-Real DR), нефропатии — риск развития альбуминурии (исследования DAIS, FIELD, ACCORD Lipid), снижать частоту развития ампутации нижних конечностей (исследования FIELD, FENO-PREVENT) и нейропатии (исследование FIELD) [140][143-146]. Такая уникальная способность фенофибрата уменьшать вероятность развития микрососудистых осложнений у пациентов с СД и ГТГ послужила поводом для применения этого препарата в профилактических целях.
Комбинация фенофибрата со статином. Ввиду доказанной целесообразности использования комбинированной терапии особого внимания заслуживают данные по безопасности совместного применения статинов и фенофибрата. Фенофибрат — единственный препарат данной группы, который можно комбинировать со статинами. Фенофибрат, в отличие от статинов, метаболизируется под действием уридинглюкуронилтрансферазы (UGT) без участия цитохромов, и пути их метаболизма не пересекаются [147]. Соответственно, совместное использование фенофибрата с различными статинами не повышает их плазменной концентрации и площади под кривой "концентрация-время" (Cmax и AUC). Значит, имеется низкая вероятность развития мышечных симптомов в сравнении, например, с высокоинтенсивной терапией статинами.
В ряде РКИ была показана эффективность комбинированной терапии статина и фибрата по снижению уровней ТГ и ХС не-ЛВП. У пациентов с гиперхолестеринемией и СД 2 типа терапия статинами в средних дозах в комбинации с фенофибратом обеспечивала пятикратное увеличение числа больных, достигавших целевых значений апоВ — белка атерогенных ТГ-богатых ЛП, ЛНП и ХС не-ЛВП [148]. В другом РКИ у пациентов с умеренно повышенным уровнем ХС ЛНП и ТГ комбинированная терапия статина и фенофибрата также содействовала большему снижению уровня ХС не-ЛВП, чем применение монотерапии статином [149].
Интересными оказались данные когортного исследования на выборке пациентов с метаболическим синдромом старше 30 лет из Корейской национальной когорты (n=39165), в котором сравнивались две тактики лечения: комбинированная терапия фенофибрата (доза 145 мг/сут.) со статином и комбинация омега-3 ПНЖК (≤2 г/сут.) со статином [150]. Клинические эффекты комбинации фенофибрата со статином были значимо лучше: RR первичной конечной точки (ИБС, ишемический инсульт, смертность от ССЗ) снижался на 21% (95% ДИ: 0,74-0,83), ИБС на 34% (0,61-0,72), ишемического инсульта на 28% (0,67-0,77) и госпитализации из-за СН на 10% (0,82-0,97). Позитивный клинический эффект (на первичную конечную точку) фенофибрата оказался сравним с эффектом омега-3 ПНЖК в группе пациентов без АССЗ и в группе пациентов, получавших омега-3 ПНЖК в дозе >2 г/сут.
Лекарственные препараты, содержащие омега-3 ПНЖК, такие как ДГК и ЭПК, применяются для лечения ГТГ. Для оптимального снижения уровня ТГ в плазме крови рекомендуется использовать большие дозы (2-4 г/сут.) этих препаратов.
Гиполипидемический механизм действия ЭПК связан: с подавлением синтеза ЛОНП в печени и ускорением выведения этих частиц из кровотока; усилением β-окисления свободных ЖК, что уменьшает количество субстрата, доступного для синтеза ТГ и ЛОНП [151]. ЭПК улучшает функцию эндотелия, повышая активность эндотелиальной синтазы оксида азота и снижая экспрессию факторов адгезии эндотелиальных клеток сосудов и хемокинов; стабилизирует фосфолипидный биослой мембран; ингибирует агрегацию тромбоцитов; проявляет противовоспалительное и антиоксидантное действия [152]. ДГК содействует снижению уровня апоСIII, что улучшает катаболизм ТГ-богатых ЛП и снижает уровень ТГ. ДГК увеличивают текучесть клеточных мембран, оказывают умеренный антиоксидантный эффект [153]. Итак, ЭПК и ДГК обладают позитивными биологическими эффектами [154]. Следует отметить, что по данным исследования DEFAT при 3-летнем наблюдении более заметное снижение уровня ТГ в плазме крови наблюдалось на фоне приема комбинации ЭПК+ДГК, чем на монопрепарате ЭПК [155].
Метаанализ, включавший 90 РКИ и 72598 пациентов, продемонстрировал практически линейное дозозависимое влияние омега-3 ПНЖК (ДГК, ЭПК или обеих) на уровни ТГ и ХС не-ЛВП в плазме крови, а также подтвердил целесообразность применения подобных препаратов в дозе >2 г/сут. [156]. Омега-3 ПНЖК снижают уровень ТГ в среднем от 25% при их исходном значении <2 ммоль/л до 34% при их значении ≥2 ммоль/л (метаанализ 36 плацебо-контролируемых перекрестных РКИ). При выраженной ГТГ омега-3 ПНЖК могут назначаться в комбинации со статинами и/или фенофибратом.
Результаты клинических исследований с применением омега-3 ПНЖК различаются между собой. Это связано с разными причинами: различным дизайном исследования; характеристикой исследуемой популяции (включая уровень ТГ в плазме крови, величину ССР пациента, применение статинов и/или другой терапии и т.д.); отсутствием иногда убедительного нивелирования межгрупповых различий; широкой гетерогенностью состава омега-3 ПНЖК (разное соотношение ЭПК и ДГК, разные формы содержания омега-3 ПНЖК: этиловые эфиры и карбоновые кислоты) и используемыми дозировками [153]. В клинических исследованиях продемонстрирована зависимость клинического эффекта омега-3 ПНЖК от их дозы [150].
В исследовании EVAPORATE (Effect of Vascepa on Improving Coronary Atherosclerosis in People With High Triglycerides Taking Statin Therapy) у 80 пациентов с высоким ССР и ГТГ, получавших статины, назначение ЭПК (эйкозапента этила) приводило к значимому уменьшению объема бляшек по данным компьютерной томографической ангиографии коронарных артерий через 18 мес. лечения в сравнении с плацебо [157].
Метаанализ всех РКИ (n=135267), изучавших влияние омега-3 ПНЖК на клинические исходы (40 РКИ; исследования STRENGTH (Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridaemia) и OMEMI (Omega-3 Fatty acids in Elderly with Myocardial Infarction) были исключены), показал, что повышение дозы омега-3 ПНЖК на 1 г/сут. снижает риск развития ССО на 5,8% (p<0,01) [158].
В японское исследование JELIS (Japan EPA Lipid Intervention Study) было включено 18645 пациентов с концентрацией общего ХС >6,5 ммоль/л и средним уровнем ТГ 1,73 ммоль/л (наблюдение 4,6 лет). В нем сравнивали клинические эффекты от применения ЭПК (в виде этилового эфира) в дозе 1,8 г/сут. в сочетании с низкоинтенсивной терапией статинами и назначение только низкоинтенсивной терапии статинов [159]. У пациентов, получавших омега-3 ПНЖК в сочетании со статинами, наблюдалось снижение частоты коронарных событий на 19% (р=0,011) по сравнению с теми, кто получал только статины; у пациентов с ранее установленными АССЗ риск ССО снижался также на 19% (р=0,048).
Наиболее важные клинические данные были получены в двойном слепом плацебо-контролируемом РКИ REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial). В исследование было включено 8179 пациентов с доказанными ССЗ или СД с наличием не менее одного дополнительного фактора риска и уровнем ТГ в плазме крови 1,52-5,63 ммоль/л [160]. Изучалось влияние 4 г эйкозапента этила, высокоочищенного препарата ЭПК, или плацебо (минеральное масло) на риск развития ССО (наблюдение 4,9 лет). У пациентов, получавших эйкозапент этил, показано снижение первичной конечной точки (сердечно-сосудистой смерти, нефатального ИМ, нефатального ишемического инсульта, реваскуляризации, нестабильной стенокардии) на 25% (95% ДИ: 0,68-0,93; р<0,001). Подробный иерархический анализ показал существенное и статистически значимое снижение случаев фатального или нефатального ИМ — на 31%, сердечно-сосудистой смерти — на 20%, фатального или нефатального инсульта — на 28% [161].
Результаты подтвердили, что у пациентов с ГТГ для снижения ССР целесообразно применять препараты омега-3 ПНЖК, содержащие в своем составе этиловые эфиры ПНЖК, а не омега-3 карбоновые кислоты (как в исследовании STRENGTH), длительно и в достаточно высоких дозах [153][162].
Данные метаанализа, включавшего 12 РКИ и 81364 пациентов с хронической СН или с ССЗ, свидетельствуют о том, что применение омега-3 ПНЖК снижает на 9% (p=0,02) число повторных госпитализаций в связи с хронической СН [163].
На хорошую переносимость и безопасность терапии препаратами омега-3 ПНЖК указывает большой опыт их длительного применения в реальной клинической практике и результаты выполненных многочисленных РКИ. Сообщается о возможном увеличении случаев фибрилляции предсердий и кровотечений при назначении омега-3 ПНЖК. В основном это относится к высокой дозе ЭПК (эйкозапенту этилу). В РКИ REDUCE-IT частота развития фибрилляции предсердий на фоне приема эйкозапента этила составляла 5,3% vs 3,9% на плацебо (р=0,003), а частота кровотечений 2,7% vs 2,1% (р=0,06), соответственно (при отсутствии фатальных кровотечений). Однако эти риски не перевешивают клинические преимущества от назначения омега-3 ПНЖК пациентам. Все случаи кровотечений были незначительными (например, кровоподтеки на коже или синяки без повышения риска фатальных, внутричерепных, желудочно-кишечных или мочеполовых кровотечений), а фибрилляция предсердий чаще развивалась у пациентов, имеющих подобные нарушения ритма сердца в анамнезе [160].
Следует отметить, что частота развития фибрилляции предсердий на фоне применения препарата ЭПК+ДГК ниже по сравнению с препаратом ЭПК (эйкозапентом этилом). Так, по результатам наблюдательного исследования совокупная частота развития фибрилляции предсердий за два года наблюдения составила 5,32% при приеме ЭПК и 3,99% при приеме комбинации ДГК+ЭПК (RR 1,242; 95% ДИ: 1,061-1,455; р=0,0032) [164].
Итак, имеются доказательства положительного влияния омега-3 ПНЖК на снижение частоты ССО при низком риске появления нежелательных эффектов. Принимая лекарственный препарат омега-3 ПНЖК желательно контролировать не только уровень ТГ, но и содержание в плазме крови ХС ЛНП. У некоторых пациентов ХС ЛНП может несколько повышаться, но без активного формирования атерогенных мелких плотных частиц ЛНП.
Для получения хорошего эффекта в реальной клинической практике важно применять рецептурные омега-3 ПНЖК (этиловые эфиры омега-3 ПНЖК), представляющие собой высококонцентрированные очищенные препараты. Продукты из рыбьего жира, отпускаемые без рецепта, классифицируются как пищевые добавки и не заменяют лекарственный препарат [14]. В составе добавок могут содержаться примеси, в т.ч. насыщенные жиры и окисленные липиды, загрязняющие вещества или другие ингредиенты, вредные для организма человека. Добавки с рыбьим жиром переносятся хуже, чем одобренные к применению лекарственные препараты омега-3 ПНЖК (в России — это лекарственный препарат Омакор). Нет доказательств того, что безрецептурные препараты с рыбьим жиром или пищевые добавки улучшают состояние сердечно-сосудистой системы. Они не рекомендуются для снижения риска АССЗ [14].
Аферез ЛП резко снижает (на 50-80%) уровень ТГ в плазме крови и, таким образом, теоретически устраняет причинный фактор панкреатита, вызванного ГТГ [165][166]. Однако уровни ТГ могут быстро восстанавливаться после плазмафереза, если первопричина ГТГ не устранена должным образом.
Наиболее эффективными методами терапевтического афереза, удаляющими ТГ из плазмы крови, являются: непрерывный плазмаферез со снижением концентрации ТГ до 48%, каскадная плазмофильтрация — до 54%, иммуносорбция ЛНП и Лп(а) (поликлональные антитела) — до 56%, гепарин ЛНП/фибриноген преципитация (HELP) — до 63%, декстран-сульфат плазмосорбции (одноразовый сорбент) — до 68% [167][168].
Применение процедур реофереза за предельно короткое время приводит к выраженному улучшению реологии и микроциркуляции крови, а также функции эндотелия сосудов. Особенно это важно для беременных женщин с ГТГ, поскольку большинство липидснижающих препаратов им противопоказано. После процедур терапевтического афереза значимо повышается чувствительность к лекарственным препаратам.
Ключевые положения
У пациентов с высоким, очень высоким и экстремальным ССР требуется регулярно измерять уровень ТГ натощак. Необходимо выяснить возможные причины ГТГ. Всем пациентам с историей острого панкреатита следует определять уровень ТГ в образцах крови, взятой натощак. Пациентам с уровнем ТГ в плазме крови >1,7 ммоль/л, особенно при наличии АССЗ, рекомендуется корректировать их содержание немедикаментозными и медикаментозными средствами. Уменьшение концентрации ТГ в плазме крови — важная составляющая снижения ССР и остаточного липидного риска после приема ХС-снижающих препаратов.
При изолированном повышении уровня ТГ от 1,7 до 2,3 ммоль/л применять немедикаментозные методы; при высоком и очень высоком ССР рассмотреть возможность назначения гипотриглицеридемических препаратов (в виде омега-3 ПНЖК этиловых эфиров 90 в дозе 2-4 г). Пациентам с комбинированной ГЛП необходимо исходно оценить уровень ХС ЛНП, снизить его до целевого значения статинами (в виде монотерапии или комбинации ХС-снижающих препаратов); при сохранении уровня ТГ в пределах от 1,7 до 2,3 ммоль/л к ХС-снижающей терапии добавить лекарственный препарат омега-3 ПНЖК. При уровне ТГ >2,3 ммоль/л целесообразно назначить фенофибрат или омега-3 ПНЖК, несмотря на терапию, направленную на снижение концентрации ХС ЛНП. Если концентрация ТГ ≥5,0 ммоль/л, то для предупреждения развития панкреатита в состав терапии целесообразно включать фенофибрат и омега-3 ПНЖК. Таких пациентов лучше направлять на консультацию в липидный центр для углубленного обследования и проведения терапевтического афереза.
Положительные результаты выполненных РКИ с применением лекарственных вмешательств, направленных на коррекцию ГТГ, дают основание для разработки инновационных терапевтических средств, снижающих уровни ТГ, ТГ-богатых ЛП и их ремнантов, с достижением потенциальной пользы для сердечно-сосудистой системы.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. WHO newsletter. Cardiovascular diseases [cited by Apr 20, 2019]. Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
2. United States Environmental Protection Agency. Guidelines for eating fish that contain mercury. Available at: https://www.epa.gov/mercury/guidelineseating-fish-contain-mercury. Accessed December, 2024; Institute of Medicine. Seafood choices: balancing benefits and risks. Available at: https://www.nap.edu/catalog/11762/seafood-choices-balancing-benefits-andrisks.Accessed December, 2024. doi:10.17226/11762.
1. Ежов М. В., Кухарчук В. В., Сергиенко И. В. и др. Нарушения липидного обмена. Клинические рекомендации 2023. Российский кардиологический журнал. 2023;28(5):5471. doi:10.15829/1560-4071-2023-5471. EDN YVZOWJ.
2. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020; 41:111-88. doi:10.1093/eurheartj/ehz455.
3. Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2021;42:3227-337. doi:10.1093/eurheartj/ ehab484.
4. Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2(8):655-66. doi:10.1016/S2213-8587(13)70191-8.
5. Langsted A, Freiberg JJ, Nordestgaard BG. Fasting and nonfasting lipid levels: influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation. 2008;118:2047-56. doi:10.1161/ CIRCULATIONAHA.108.804146.
6. Nordestgaard BG, Langsted A, Mora S, et al. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points — a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J. 2016;37(25):1944-58. doi:10.1093/eurheartj/ehw152.
7. Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease New Insights From Epidemiology, Genetics, and Biology. Circ Res. 2016;118:547-63. doi:10.1161/CIRCRESAHA.115.306249.
8. Ginsberg HN, Packard CJ, Chapman MJ, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42(47):4791-806. doi:10.1093/eurheartj/ehab551.
9. Castelli WP. Epidemiology of triglycerides: a view from Framingham. American J Cardiol. 1992;70(19):H3-H9. doi:10.1016/0002-9149(92)91083-g.
10. Carr RA, Rejowski BJ, Cote GA, et al. Systematic review of hypertriglyceridemiainduced acute pancreatitis: a more virulent etiology? Pancreatology. 2016;16:469-76. doi:10.1016/j.pan.2016.02.011.
11. Pedersen SB, Langsted A, Nordestgaard BG. Nonfasting mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis. JAMA Intern Med. 2016;176:1834-42. doi:10.1001/jamainternmed.2016.6875.
12. Murphy M, Shemg X, MacDonald TM. Hypertriglyceridemia and acute pancreatitis. JAMA Intern Med. 2013;173(2):162-4. doi:10.1001/2013.jamaintermed.477.
13. Faghih M, Singh VK. Do elevated triglycerides truly trigger acute pancreatitis? Dig Dis Sci. 2019;64:616-8. doi:10.1007/s10620-019-05501-0.
14. Virani SS, Morris PB, Agarwala A, et al. 2021 ACC expert consensus decision pathway on the management of ASCVD risk reduction in patients with persistent hypertriglyceridemia: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2021;78(9):960-93. doi:1016/j.jacc.2021.06.011.
15. Stürzebecher PE, Katzmann JL, Laufs U. What is ‘remnant cholesterol'? Eur Heart J. 2023;44:1446-8. doi:10.1093/eurheartj/ehac783.
16. Remaley AT, Otvos JD. Methodological issues regarding: "A third of nonfasting plasma cholesterol is in remnant lipoproteins: lipoprotein subclass profiling in 9293 individuals". Atherosclerosis. 2020;302:55-6. doi:10.1016/j.atherosclerosis.2020.01.020.
17. Tybjærg-Hansen A, Nordestgaard BG, Christoffersen M. Triglyceride-rich remnant lipoproteins are more atherogenic than LDL per particle: is this important? Eur Heart J. 2023;44:4196-8. doi:10.1093/eurheartj/ehad419.
18. White KT, Moorthy MV, Akinkuolie AO, et al. Identifying an optimal cutpoint for the diagnosis of hypertriglyceridemia in the nonfasting state. Clin Chem. 2015;61:1156-63. doi:10.1373/clinchem.2015.241752.
19. Raja V, Aguiar C, Alsayed N, et al. Non-HDL-cholesterol in dyslipidemia: Review of the state-of-the-art literature and outlook. Atherosclerosis. 2023;383:117312. doi:10.1016/j.atherosclerosis.2023.117312.
20. Fredrickson DS. An international classification of hyperlipidemias and hyperlipoproteinemias. Ann Intern Med. Ann Intern Med. 1971;75(3):471-2. doi:10.7326/0003-481975-3-471.
21. Gill PK, Dron JS, Berberich AJ, et al. Combined hyperlipidemia is genetically similar to isolated hypertriglyceridemia. J Clin Lipidol. 2021;15(1):79-87. doi:10.1016/j.jacl.2020.11.006.
22. Laufs U, Parhofer KG, Ginsberg HN, Hegele RA. Clinical review on triglycerides. Eur Heart J. 2020;41:99-109. doi:10.1093/eurheartj/ehz785.
23. Cohen JD, Cziraky MJ, Cai Q, et al. 30-year trends in serum lipids among United States adults: results from the National Health and Nutrition Examination Surveys II, III, and 19992006. Am J Cardiol. 2010;106(7):969-75. doi:10.1016/j.amjcard.2010.05.030.
24. Simha V. Management of hypertriglyceridemia. BMJ. 2020;371:m3109. doi:10.1136/bmj. m3109.
25. Сhyzhyk V, Kozmic S, Brown AS, et al. Extreme hypertriglyceridemia: genetic diversity, pancreatitis, pregnancy, and prevalence. J Clin Lipidol. 2019;13(1):89-99. doi:10.1016/ j.jacl.2018.09.007.
26. Gitt AK, Drexel H, Feely J, et al. DYSIS Investigators. Persistent lipid abnormalities in statin-treated patients and predictors of LDL-cholesterol goal achievement in clinical practice in Europe and Canada. Eur J Prev Cardiol. 2012;19(2):221-30. doi:10.1177/1741826711400545.
27. Драпкина О. М., Имаева А. Э., Куценко В. А. и др. Дислипидемии в Российской Федерации: популяционные данные, ассоциации с факторами риска. Кардиоваскулярная терапия и профилактика. 2023;22(8S):3791. doi:10.15829/1728-8800-2023-3791. EDN DGYJLA.
28. Karpov Y, Khomitskaya Y. PROMETHEUS: an observational, cross-sectional, retrospective study of hypertriglyceridemia in Russia. Cardiovasc Diabetol. 2015;14:115. doi:10.1186/ s12933-015-0268-2.
29. Ghandehari H, Kamal-Bahl S, Wong ND. Prevalence and extent of dyslipidemia and recommended lipid levels in US adults with and without cardiovascular comorbidities: The National Health and Nutrition Examination Survey 2003-2004. Am Heart J. 2008;156(1):112-9. doi:10.1016/j.ahj.2008.03.005.
30. Chait A, Eckel RH. The chylomicronemia syndrome is most often multifactorial: a narrative review of causes and treatment. Ann Intern Med. 2019;170:626-34. doi:10.7326/ M19-0203.
31. Gugliucci A. The chylomicron saga: time to focus on postprandial metabolism. Front. Endocrinol. 2023;14:1322869. doi:10.3389/fendo.2023.1322869.
32. Nordestgaard BG, Zilversmit DB. Large lipoproteins are excluded from the arterial wall in diabetic cholesterol-fed rabbits. J Lipid Res. 1988;29:1491-500.
33. Бубнова М. Г., Оганов Р. Г. Нарушенная толерантность к пищевым жирам и ее значение в атеротромбогенезе. Терапевтический архив. 2004,1:73-8.
34. Sylvers-Davie KL, Davies BSJ. Regulation of lipoprotein metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am J Physiol Endocrinol Metab. 2021;321(4):E493-508. doi:10.1152/ajpendo.00195.2021.
35. Williams KJ, Chen K. Recent insights into factors affecting remnant lipoprotein uptake. Curr Opin Lipidol. 2010;21(3):218-28. doi:10.1097/MOL.0b013e328338cabc.
36. Veniant MM, Zlot CH, Walzem RL, et al. Lipoprotein clearance mechanisms in LDL receptor-deficient "Apo-B48-only" and "Apo-B100-only" mice. J Clin Invest. 1998;102(8): 1559-68. doi:10.1172/JCI4164.
37. Salinas CAA, Chapman MJ. Remnant lipoproteins: are they equal to or more atherogenic than LDL? Curr Opin Lipidol. 2020;31:132-9. doi:10.1097/MOL.0000000000000682.
38. Botham K, Bravo E, Elliott J, Wheeler-Jones C. Direct interaction of dietary lipids carried in chylomicron remnants with cells of the artery wall: implications for atherosclerosis development. Curr Pharm Des. 2005;11(28):3681-95. doi:10.2174/138161205774580732.
39. Dalla-Riva J, Garonna E, Elliott J, et al. Botham KM, Wheeler-Jones CP. Endothelial cells as targets for chylomicron remnants. Atheroscler Suppl. 2010;11(1):31-7. doi:10.1016/j.atherosclerosissup.2010.04.001.
40. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017;117:7-18. doi:10.1160/TH16-08-0593.
41. Pal S, Semorine K, Watts GF, Mamo J. Identification of lipoproteins of intestinal origin in human atherosclerotic plaque. Clin Chem Lab Med. 2003;41:792-5. doi:10.1515/CCLM.2003.120.
42. Rapp JH, Lespine A, Hamilton RL, et al. Triglyceride-rich lipoproteins isolated by selected-affinity anti-apolipoprotein B immunosorption from human atherosclerotic plaque. Arterioscler Thromb. 1994;14:1767-74. doi:10.1161/01.atv.14.11.1767.
43. Rosenson RS, Davidson MH, Hirsh BJ, et al. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardio-vascular disease. J Am Coll Cardiol. 2014;64:252540. doi:10.1016/j.jacc.2014.09.042.
44. Nakano T, Nakajima K, Niimi M, et al. Detection of apolipoproteins B-48 and B-100 carrying particles in lipoprotein fractions extracted from human aortic atherosclerotic plaques in sudden cardiac death cases. Clin Chim Acta. 2008;390:38-43. doi:10.1016/j.cca.2007.12.012.
45. Davidson MH. Triglyceride-rich lipoprotein cholesterol (TRL-C): the ugly stepsister of LDL-C. Eur Heart J. 2018;39:620-2. doi:10.1093/eurheartj/ehx741.
46. Örni K, Lehti S, Sjövall P, Kovanen PT. Triglyceride-rich lipoproteins as a source of proinflammatory lipids in the arterial wall. Curr Med Chem. 2018;26(9):1701-10. doi:10.2774/0929867325666180530094819.
47. Varbo A, Benn M, Tybjærg-Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298-309. doi:10.1161/CIRCULATIONAHA.113.003008.
48. Raposeiras-Roubin S, Rosselló X, Oliva B, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol. 2021;77:3031-41. doi:10.1016/j.jacc.2021.04.059.
49. Lin A, Nerlekar N, Rajagopalan A, et al. Remnant cholesterol and coronary atherosclerotic plaque burden assessed by computed tomography coronary angiography. Atherosclerosis. 2029;284:24-30. doi:10.1016/j.atherosclerosis.2019.02.019.
50. Бубнова М. Г., Аронов Д. М., Перова Н. В., Мазаев В. П. Связь уровня липемии после жировой нагрузки со степенью выраженности атеросклероза коронарных артерий. Терапевтический архив. 2004;76(6):62-7.
51. Elshazly MB, Mani P, Nissen S, et al. Remnant cholesterol, coronary atheroma progression and clinical events in statin-treated patients with coronary artery disease. Eur J Prev Cardiol. 2020;27:1091-100. doi:10.1177/2047487319887578.
52. Krauss RM, Williams PT, Brensike J, et al. Intermediate-density lipoproteins and progression of coronary artery disease in hypercholesterolemic men. Lancet. 1987;2:62-6. doi:10.1016/s0140-6736(87)92734-6.
53. Karpe F, Steiner G, Uffelman K, et al. Postprandial lipoproteins and progression of coronary atherosclerosis. Atherosclerosis. 1994;106:83-97. doi:10.1016/00219150(94)90085-x.
54. Phillips NR, Water D, Havel RJ. Plasma lipoproteins and progression of coronary artery disease evaluated by angiography and clinical events. Circulation. 1993;88:2762-70. doi:10.1161/01.cir.88.6.2762.
55. Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313-30. doi:10.1093/eurheartj/ehz962.
56. Borén J, Matikainen N, Adiels M, Taskinen MR. Postprandial hypertriglyceridemia as a coronary risk factor. Clin Chim Acta. 2014;431:131-42. doi:10.1016/j.cca.2014.01.015.
57. Björnson E, Packard CJ, Adiels M, et al. Investigation of human apoB48 metabolism using a new, integrated non-steady-state model of apoB48 and apoB100 kinetics. J Intern Med. 2019;285:562-77. doi:10.1111/joim.12877.
58. Austin MA. Plasma triglyceride and coronary heart disease. Arterioscler Thromb. 1991; 11:2-14. doi:10.1161/01.atv.11.1.2.
59. Stavenow L, Kjellström T. Influence of serum triglyceride levels on the risk for myocardial infarction in 12,510 middle aged males: interaction with serum cholesterol. Atherosclerosis. 1999;147(2):243-7. doi:10.1016/s0021-9150(99)00190-2.
60. Austin MA, Hokanson JE, Edwards KL. Hypertriglyceridemia as a cardiovascular risk factor. Am J Cardiol. 1998;81:7B-12B. doi:10.1016/s0002-9149(98)00031-9.
61. Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115(4):450-8. doi:10.1161/CIRCULATIONAHA.106.637793.
62. Di Angelantonio E, Sarwar N, Perry P, et al. Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993-2000. doi:10.1001/jama.2009.1619.
63. Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626-35. doi:10.1016/S0140-6736(14)61177-6.
64. Varbo A, Nordestgaard BG. Remnant cholesterol and risk of ischemic stroke in 112,512 individuals from the general population. Ann Neurol. 2019;85:550-9. doi:10.1002/ana.25432.
65. Ference BA, Kastelein JJP, Ray KK, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA. 2019;321:364-73. doi:10.1001/jama.2018.20045
66. Kalthoft M, Langsted A, Nordestgaard BG. Triglycerides and remnant cholesterol associated with risk of aortic valve stenosis: Mendelian randomization in the Copenhagen General Population Study. Eur Heart J. 2020;41:2288-99. doi:10.1093/eurheartj/ehaa172.
67. Park H-B, Arsanjani R, Hong S-J, et al. Impact of hypertriglyceridaemia on cardiovascular mortality according to low-density lipoprotein cholesterol in a 15.6-million population. Eur J Prevent Cardiol. 2024;31:280-90. doi:10.1093/eurjpc/zwad330.
68. Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3:213-9.
69. Patel A, Barzi F, Jamrozik K, et al. Asia Pacific Cohort Studies Collaboration. Serum triglycerides as a risk factor for cardiovascular diseases in the Asia-Pacific region. Circulation. 2004;110:2678-86. doi:10.1161/01.CIR.0000145615.33955.83.
70. Arca M, Veronesi C, D'Erasmo L, et al. Association of Hypertriglyceridemia with All-Cause Mortality and Atherosclerotic Cardiovascular Events in a Low-Risk Italian Population: The TG-REAL Retrospective Cohort Analysis. J Am Heart Assoc. 2020;9:e015801. doi:10.1161/JAHA.119.015801.
71. Patel RS, Pasea L, Soran H, et al. Elevated plasma triglyceride concentration and risk of adverse clinical outcomes in 1.5 million people: a CALIBER linked electronic health record study. Cardiovasc Diabetol. 2022;21(1):102. doi:10.1186/s12933-02201525-5.
72. Lee H, Park J-B, Hwang I-C, et al. Association of four lipid components with mortality, myocardial infarction, and stroke in statin-naïve young adults: A nationwide cohort study. Eur J Prevent Cardiol. 2020;27(8):870-81. doi:10.1177/2047487319898571.
73. Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids commonly present in young adults and coronary calcium later in life: the CARDIA (Coronary Artery Risk Development in Young Adults) study. Ann Intern Med. 2010;153(3):137-46. doi:10.7326/0003-4819-153-3-201008030-00004.
74. Jeppesen J, Hein HO, Suadicani P, et al. Relation of high TG-low HDL cholesterol and LDL cholesterol to the incidence of ischemic heart disease. An 8-year follow-up in the Copenhagen Male Study. Arterioscler Thromb Vasc Biol. 1997;17(6):1114-20. doi:10.1161/01.atv.17.6.1114.
75. Kivioja R, Pietila A, Martinez-Majander N, et al. Risk factors for early-onset ischemic stroke: a case-control study. J Am Heart Assoc. 2018;7(21):e009774. doi:10.1161/JAHA.118.009774.
76. Nordestgaard BG, Benn M, Schnohr P, Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299-308. doi:10.1001/jama.298.3.299.
77. Freiberg JJ, Tybjærg-Hansen A, Jensen JS, Nordestgaard BG. Nonfasting triglycerides and risk of ischemic stroke in the general population. JAMA. 2008;300(18):2142-52. doi:10.1001/jama.2008.621.
78. Bansal S, Buring JE, Rifai N, et al. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298(3):309-16. doi:10.1001/jama.298.3.309.
79. Varbo A, Nordestgaard BG. Nonfasting triglycerides, low-density lipoprotein cholesterol, and heart failure risk: two cohort studies of 113 554 individuals. Arterioscler Thromb Vasc Biol. 2018;38(2):464-72. doi:10.1161/ATVBAHA.117.310269.
80. Toth PP, Sephy P, Hull M, Granowitz C. Elevated Triglycerides (≥150 mg/dL) and High Triglycerides (200-499 mg/dL) Are Significant Predictors of New Heart Failure Diagnosis: A Real-World Analysis of High-Risk Statin-Treated Patients. Vascular Health and Risk Management. 2019;15:533-8. doi:10.2147/VHRM.S221289.
81. Varbo A, Benn M, Tybjærg-Hansen A, et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61(4):427-36. doi:10.1016/j.jacc.2012.08.1026.
82. Goliasch G, Wiesbauer F, Blessberger H, et al. Premature myocardial infarction is strongly associated with increased levels of remnant cholesterol. J Clin Lipidol. 2015;9:801-6. doi:10.1016/j.jacl.2015.08.009.
83. Varbo A, Freiberg JJ, Nordestgaard BG. Extreme nonfasting remnant cholesterol vs extreme LDL cholesterol as contributors to cardiovascular disease and all-cause mortality in 90000 individuals from the general population. Clin Chem. 2015;61(3):533-43. doi:10.1373/clinchem.2014.234146.
84. Jepsen AM, Langsted A, Varbo A, et al. Increased remnant cholesterol explains part of residual risk of all-cause mortality in 5414 patients with ischemic heart disease. Clin Chem. 2016;62(4):593-604. doi:10.1373/clinchem.2015.253757.
85. Bittencourt MS, Santos RD, Staniak H, et al. Relation of fasting triglyceride-rich lipoprotein cholesterol to coronary artery calcium score (from the ELSA-Brasil Study). Am J Cardiol. 2017;119(9):1352-8. doi:10.1016/j.amjcard.2017.01.033.
86. Doi T, Langsted A, Nordestgaard BG. Dual elevated remnant cholesterol and C-reactive protein in myocardial infarction, atherosclerotic cardiovascular disease, and mortality. Atherosclerosis. 2023;379:117141. doi:10.1016/j.atherosclerosis.2023.05.010.
87. Quispe R, Martin SS, Michos ED, et al. Remnant cholesterol predicts cardiovascular disease beyond LDL and ApoB: a primary prevention study. Eur Heart J. 2021;42:4324-32. doi:10.1093/eurheartj/ehab432.
88. Lee SJ, Kim S-E, Go T-H, et al. Remnant cholesterol, low-density lipoprotein cholesterol, and incident cardiovascular disease among Koreans: a national population-based study. Eur J Prevent Cardiol. 2023;30:1142-50. doi:10.1093/eurjpc/zwad036.
89. Wadström BN, Wulff AB, Pedersen KM, et al. Elevated remnant cholesterol increases the risk of peripheral artery disease, myocardial infarction, and ischaemic stroke: a cohortbased study. Eur Heart J. 2022;43:3258-69. doi:10.1093/eurheartj/ehab705.
90. Yang XH, Zhang BL, Cheng Y, et al. Association of remnant cholesterol with risk of cardiovascular disease events, stroke, and mortality: A systemic review and meta-analysis Atherosclerosis. 2023;371:21-31. doi:10.1016/j.atherosclerosis.2023.03.012.
91. Cordero A, Alvarez-Alvarez B, Escribano D, et al. Remnant cholesterol in patients admitted for acute coronary syndromes. Eur J Prevent Cardiol. 2023;30:340-8. doi:10.1093/eurjpc/zwac286.
92. Langsted A, Freiberg JJ, Tybjaerg-Hansen A, et al. Nonfasting cholesterol and triglycerides and association with risk of myocardial infarction and total mortality: the Copenhagen City Heart Study with 31 years of follow-up. J Intern Med. 2011;270:65-75. doi:10.1111/j.1365-2796.2010.02333.x.
93. Wadström BN, Pedersen KM, Wulff AB, et al. Elevated remnant cholesterol, plasma triglycerides, and cardiovascular and non-cardiovascular mortality. Eur. Heart J. 2023; 44:1432-45. doi:10.1093/eurheartj/ehac822.
94. Jørgensen AB, Frikke-Schmidt R, West AS, et al. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J. 2013;34:1826-33. doi:10.1093/eurheartj/ehs431.
95. Chapman MJ, Ginsberg HN, Amarenco P, et al. the European Atherosclerosis Society Consensus Panel. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345-61. doi:10.1093/eurheartj/ehr112.
96. Boullart AC, de Graaf J, Stalenhoef AF. Serum triglycerides and risk of cardiovascular disease. Biochim Biophys Acta. 2012;1821:867-75. doi:10.1016/j.bbalip.2011.10.002.
97. Björnson E, Adiels M, Taskinen M-R, et al. Triglyceride-rich lipoprotein remnants, lowdensity lipoproteins, and risk of coronary heart disease: a UK Biobank study. Eur Heart J. 2023;44:4186-95. doi:10.1093/eurheartj/ehad337.
98. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102-6. doi:10.1038/nature13917.
99. Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371: 32-41. doi:10.1056/NEJMoa1308027.
100. Khera AV, Won HH, Peloso GM, et al. Myocardial Infarction Genetics Consortium, DiscovEHR Study Group, CARDIoGRAM Exome Consortium, and Global Lipids Genetics Consortium. Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317:937-46. doi:10.1001/jama.2017.0972.
101. Stitziel NO, Khera AV, Wang X, et al. PROMIS and Myocardial Infarction Genetics Consortium Investigators. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054-63. doi:10.1016/j.jacc.2017.02.030.
102. Dewey FE, Gusarova V, O'Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123-33. doi:10.1056/NEJMoa1510926.
103. Stitziel NO, Stirrups KE, Masca NG, et al. Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators; Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374:1134-44. doi:10.1056/NEJMoa1507652.
104. Helkkula P, Kiiskinen T, Havulinna AS, et al. ANGPTL8 protein-truncating variant associated with lower serum triglycerides and risk of coronary disease. PLoS Genet. 2021; 17:e1009501. doi:10.1371/journal.pgen.1009501.
105. Thomsen M, Varbo A, Tybjaerg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem. 2014; 60:737-46. doi:10.1373/clinchem.2013.219881.
106. Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22-31. doi:10.1056/NEJMoa1307095.
107. Dhindsa DS, Sandesara PB, Shapiro MD, Wong ND. The evolving understanding and approach to residual cardiovascular risk management. Front Cardiovasc Med. 2020;7:88. doi:10.3389/fcvm.2020.00088.
108. Toth Peter P, Fazio S, Wong ND. Risk of cardiovascular events in patients with hypertriglyceridaemia: A review of real-world evidence Diabetes Obes Metab. 2020;22:279-89. doi:10.1111/dom.13921.
109. Marston NA, Giugliano RP, Im KAh, et al. Association between triglyceride lowering and reduction of cardiovascular risk across multiple lipid-lowering therapeutic classes: a systematic review and meta-regression analysis of randomized controlled trials. Circulation. 2019;140:1308-17. doi:10.1161/CIRCULATIONAHA.119.041998.
110. Schwartz GG, Abt M, Bao W, et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol. 2015;65(21):2267-75. doi:10.1016/j.jacc.2015.03.544.
111. Sacks FM, Tonkin AM, Shepherd J, et al. Effect of pravastatin on coronary disease events in subgroups defined by coronary risk factors: the Prospective Pravastatin Pooling Project. Circulation. 2000;102:1893-900. doi:10.1161/01.cir.102.16.1893.
112. Miller M, Cannon CP, Murphy SA, et al. PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724-30. doi:10.1016/j.jacc.2007.10.038.
113. Faergeman O, Holme I, Fayyad R, et al. Steering Committees of IDEAL and TNT Trials. Plasma triglycerides and cardiovascular events in the Treating to New Targets and Incremental Decrease in End-Points through Aggressive Lipid Lowering trials of statins in patients with coronary artery disease. Am J Cardiol. 2009;104(4):459-63. doi:10.1016/j.amjcard.2009.04.008.
114. Anderson JW, Konz EC. Obesity and disease management: effects of weight loss on comorbid conditions. Obes Res. 2001;9(Suppl 4):326S-334S. doi:10.1038/oby.2001.138.
115. Couillard C, Després JP, Lamarche B, et al. Effects of endurance exercise training on plasma HDL cholesterol levels depend on levels of triglycerides: evidence from men of the Health, Risk Factors, Exercise Training and Genetics (HERITAGE) Family Study. Arterioscler Thromb Vasc Biol. 2001;21(7):1226-32. doi:10.1161/hq0701.092137.
116. Бубнова М. Г., Аронов Д. М., Олферьев А. М., Бондаренко И. З. Модификация уровней липопротеидов и аполипопротеинов крови с помощью физических нагрузок разного вида и интенсивности у здоровых мужчин с нормои гиперлипидемией. Кардиоваскулярная терапия и профилактика. 2005;4(2):74-83.
117. Aronov DM, Bubnova MG, Perova NV, et al. The effect of maximal versus submaximal exertion on postprandial lipid levels in individuals with and without coronary heart disease. J Clin Lipidol. 2017;11:369-76. doi:10.1016/j.jacl.2017.01.007.
118. Kirkpatrick CF, Sikand G, Petersen KS, et al. Nutrition interventions for adults with dyslipidemia: A Clinical Perspective from the National Lipid Association. J Clin Lipidol. 2023;17(4):428-51. doi:10.1016/j.jacl.2023.05.099.
119. Williams L, Rhodes KS, Karmally W, et al. Familial chylomicronemia syndrome: Bringing to life dietary recommendations throughout the life span. J Clin Lipidol. 2018;12(4): 908-19. doi:10.1016/j.jacl.2018.04.010.
120. Fechner E, Smeets ETHC, Schrauwen P, Mensink RP. The Effects of Different Degrees of Carbohydrate Restriction and Carbohydrate Replacement on Cardiometabolic Risk Markers in Humans-A Systematic Review and Meta-Analysis. Nutrients. 2020;12(4):991. doi:10.3390/nu12040991.
121. Stoernell CK, Tangney CC, Rockway SW. Short-term changes in lipoprotein subclasses and C-reactive protein levels of hypertriglyceridemic adults on low-carbohydrate and lowfat diets. Nutr Res. 2008;28(7):443-9. doi:10.1016/j.nutres.2008.03.013.
122. Wycherley TP, Moran LJ, Clifton PM, et al. Effects of energy-restricted high-protein, low-fat compared with standard-protein, low-fat diets: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;96(6):1281-98. doi:10.3945/ajcn.112.044321.
123. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucosesweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119(5):1322-34. doi:10.1172/JCI37385.
124. Kodama S, Horikawa C, Fujihara K, et al. Relationship between intake of fruit separately from vegetables and triglycerides — A meta-analysis. Clin Nutr ESPEN. 2018;27:53-8. doi:10.1016/j.clnesp.2018.07.001.
125. Leslie MA, Cohen DJA, Liddle DM, et al. A review of the effect of omega-3 polyunsaturated fatty acids on blood triacylglycerol levels in normolipidemic and borderline hyperlipidemic individuals. Lipids Health Dis. 2015;14:53. doi:10.1186/s12944-015-0049-7.
126. Liu YX, Yu JH, Sun JH, et al. Effects of Omega-3 Fatty Acids Supplementation on Serum Lipid Profile and Blood Pressure in Patients with Metabolic Syndrome: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Foods. 2023;12(4):725. doi:10.3390/foods12040725.
127. Khorshidi M, Hazaveh ZS, Alimohammadi-kamalabadi M, et al. Effect of omega-3 supplementation on lipid profile in children and adolescents: a systematic review and meta-analysis of randomized clinical trials. Nutr J. 2023;22:9. doi:10.1186/s12937-02200826-5.
128. Jones PH, Davidson MH, Stein EA, et al.; STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR Trial). Am J Cardiol. 2003;92(2):152-60. doi:10.1016/s00029149(03)00530-7.
129. Арутюнов Г. П., Бойцов С. А., Воевода М. И. и др. Коррекция гипертриглицеридемии с целью снижения остаточного риска при заболеваниях, вызванных атеросклерозом. Заключение Совета экспертов Российского кардиологического общества, Российского научного медицинского общества терапевтов, Евразийской ассоциации терапевтов, Национального общества по изучению атеросклероза, Российской ассоциации эндокринологов и Национальной исследовательской лиги кардиологической генетики. Рациональная Фармакотерапия в Кардиологии. 2019;15(2):282-8. doi:10.20996/1819-6446-2019-15-2-282-288.
130. Staels B, Dallongeville J, Auwerx J, et al. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation. 1998;98:2088-93. doi:10.1161/01.cir.98.19.2088.
131. Kim NH, Kim SG. Fibrates Revisited: Potential Role in Cardiovascular Risk Reduction Diabetes Metab J. 2020;44:213-21. doi:10.4093/dmj.2020.0001.
132. Fruchart JC, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today (Barc). 2006;42(1):39-64. doi:10.1358/dot.2006.42.1.963528.
133. Keating GM, Croom KF. Fenofibrate: a review of its use in primary dyslipidaemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs. 2007;67(1):121-53. doi:10.2165/00003495-200767010-00013.
134. Feher MD, Caslake M, Foxton J, et al. Atherogenic lipoprotein phenotype in type 2 diabetes: reversal with micronised fenofibrate. Diabetes Metab Res Rev. 1999;15:395. doi:10.1002/(SICI)1520-7560(199911/12)15:6<395:AID-DMRR65>3.0.CO;2-N.
135. Ezhov MV, Arutyunov GP. Effectiveness and Safety of Fenofibrate in Routine Treatment of Patients with Hypertriglyceridemia and Metabolic Syndrome. Diseases. 2023;11:140. doi:10.3390/diseases11040140.
136. DAIS investigators. Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet. 2001;357:905-10. doi:10.1016/S0140-6736(00)04209-4.
137. Keech A, Simes RJ, Barter P, et al. The FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-61. doi:10.1016/S0140-6736(05)67667-2.
138. Ginsberg HN, Elam MB, Lovato LС, et al. The ACCORD Study Group. Effects of Combination Lipid Therapy in Type 2 Diabetes Mellitus. N Engl J Med. 2010;362:1563-74. doi:10.1056/NEJMoa1001282.
139. Wierzbicki AS. FIELD of dreams, fields of tears: a perspective on the fibrate trials. Int J Clin Pract. 2006;60(4):442-9. doi:10.1111/j.1368-5031.2006.00882.x.
140. Jo SH, Nam H, Lee J, Park S, et al. Fenofibrate Use Is Associated With Lower Mortality and Fewer Cardiovascular Events in Patients With Diabetes: Results of 10,114 Patients From the Korean National Health Insurance Service CohortDiabetes Care. 2021;44:186876. doi:10.2337/dc20-1533.
141. Kim NH, Han KH, Choi J, et al. Use of fenofibrate on cardiovascular outcomes in statin users with metabolic syndrome: propensity matched cohort study. BMJ. 2019;366:5125. doi:10.1136/bmj.l5125.
142. Das Pradhan A, Glynn RJ, Fruchart JC, et al. PROMINENT Investigators. Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N Engl J Med. 2022;387:1923-34. doi:10.1056/NEJMoa2210645.
143. Ku EJ, Kim B, Han K, et al. Fenofibrate to prevent amputation and reduce vascular complications in patients with diabetes: FENO-PREVENT. Cardiovascular Diabetology. 2024;23:329. doi:10.1186/s12933-024-02422-9.
144. Elam M, Lovato L, Ginsberg H. The ACCORD-Lipid study: implications for treatment of dyslipidemia in Type 2 diabetes mellitus. Clin Lipidol. 2011;6(1):9-20. doi:10.2217/clp.10.84.
145. Li J, Shi L, Zhao G, et al. High triglyceride levels increase the risk of diabetic microvascular complications: a cross-sectional study. Lipids Health Dis. 2023;22(1):109. doi:10.1186/s12944-023-01873-5.
146. Gitay MN, Sohail A, Arzoo Y, Shakir MA. Changes in serum lipids with the onset and progression of Diabetic Retinopathy in Type-II Diabetes Mellitus. Pak J Med Sci. 2023; 39(1):188-91. doi:10.12669/pjms.39.1.6265.
147. Franssen R, Vergeer M, Stroes ES, Kastelein JJ. Combination statin-fibrate therapy: safety aspects. Diabetes Obes Metab. 2009;11(2):89-94. doi:10.1111/j.1463-1326.2008.00917.х.
148. Jones PH, Cusi K, Davidson MH, et al. Efficacy and safety of fenofibric acid co-administered with lowor moderate-dose statin in patients with mixed dyslipidemia and type 2 diabetes mellitus: results of a pooled subgroup analysis from three randomized, controlled, double-blind trials. Am J Cardiovasc Drugs. 2010;10(2):73-84. doi:10.2165/10061630000000000-00000.
149. Roth EM, McKenney JM, Kelly MT, et al. Efficacy and safety of rosuvastatin and fenofibric acid combination therapy versus simvastatin monotherapy in patients with hypercholesterolemia and hypertriglyceridemia: a randomized, double-blind study. Am J Cardiovasc Drugs. 2010;10(3):175-86. doi:10.2165/11533430-000000000-00000.
150. Kim NH, Kim JY, Choi J, Kim SG. Associations of omega-3 fatty acids vs. fenofibrate with adverse cardiovascular outcomes in people with metabolic syndrome: propensity matched cohort study. Eur Heart J — Cardiovascular Pharmacotherapy. 2024;10(2):118-
151. doi:10.1093/ehjcvp/pvad090.
152. Oscarsson J, Hurt-Camejo E. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and their mechanisms of action on apolipoprotein B-containing lipoproteins in humans: a review. Lipids in Health and Disease. 2017;16(1):149. doi:10.1186/s12944-017-0541-3.
153. Drexel H, Tamargo J, Kaski JC, et al. Triglycerides revisited: is hypertriglyceridaemia a necessary therapeutic target in cardiovascular disease? Eur Heart J — Cardiovascular Pharmacotherapy. 2023;9(6):570-82. doi:10.1093/ehjcvp/pvad044.
154. Kaur Gurleen, Mason RP, Steg PhG, Bhatt DL. Omega-3 fatty acids for cardiovascular event lowering. Eur J Prevent Cardiol. 2024;31:1005-14. doi:10.1093/eurjpc/zwae003.
155. Banaszak M, Dobrzyńska M, Kawka A, et al. Role of Omega-3 fatty acids eicosapentaenoic (EPA) and docosahexaenoic (DHA) as modulatory and anti-inflammatory agents in noncommunicable diet-related diseases — Reports from the last 10 years. Clin Nutr ESPEN. 2024;63:240-58. doi:10.1016/j.clnesp.2024.06.053.
156. Sezai A, Unosawa S, Taoka M, et al. Long-Term Comparison of Ethyl Icosapentate vs. Omega-3-Acid Ethyl in Patients With Cardiovascular Disease and Hypertriglyceridemia (DEFAT Trial). Circ J. 2019;83(6):1368-76. doi:10.1253/circj.CJ-18-076.
157. Wang T, Zhang X, Zhou N, et al. Association between omega-3 fatty acid intake and dyslipidemia: a continuous dose-response meta-analysis of randomized controlled trials. J Am Heart Assoc. 2023;12(11):e029512. doi:10.1161/JAHA.123.029512.
158. Budoff MJ, Muhlestein JB, Bhatt DL, et al. Effect of icosapent ethyl on progression of coronary atherosclerosis in patients with elevated triglycerides on statin therapy: a prospective, placebo-controlled randomized trial (EVAPORATE): interim results. Cardiovascular Research. 2021;117(4):1070-7. doi:10.1093/cvr/cvaa184.
159. Bernasconi AA, Wiest MM, Lavie CJ, et al. Effect of Omega-3 Dosage on Cardiovascular Outcomes: An Updated Meta-Analysis and Meta-Regression of Interventional Trials. Mayo Clinic Proceedings. 2021;96(2):304-13. doi:10.1016/j.mayocp.2020.08.034.
160. Yokoyama M, Origasa H, Matsuzaki M, et al. Japan EPA lipid intervention study (JELIS) Investigators. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369(9567):1090-8. doi:10.1016/S0140-6736(07)60527-3.
161. Bhatt DL, Steg PG, Miller M, et al.; REDUCE-IT Investigators. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380(1):11-22. doi:10.1056/NEJMoa1812792.
162. Bhatt DL, Steg PG, Miller M, et al. REDUCE-IT Investigators. Effects of Icosapent Ethyl on Total Ischemic Events: From REDUCE-IT. J Am Coll Cardiol. 2019;73(22):2791-802. doi:10.1016/j.jacc.2019.02.032.
163. Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of high-dose Omega-3 fatty acids vs corn oil on Major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA. 2020;324:2268-80. doi:10.1001/ jama.2020.22258.
164. Barbarawi M, Lakshman H, Barbarawi O, et al. Omega-3 supplementation and heart failure: A meta-analysis of 12 trials including 81,364 participants. Contemporary Clinical Trials. 2021;107:106458. doi:10.1016/j.cct.2021.106458.
165. Sarker J, Kim M, Munger MA, Kim K. Icosapent Ethyl-Associated New Atrial Fibrillation Incidence compared to Omega-3 Fatty Acids: An Observational Cohort Study. Circulation. 2024;150(Suppl 1):A4140072-A4140072. doi:10.1101/2024.09.16.24313779.
166. Valdivielso P, Ramirez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med. 2014;25:689-94. doi:10.1016/j.ejim.2014.08.008.
167. Berberich AJ, Ziada A, Zou GY, Hegele RA. Conservative management in hypertriglyceridemia-associated pancreatitis. J Intern Med. 2019;286:644-50. doi:10.1111/joim.12925.
168. Konovalov GA, Filonenko IV, Akopyan VS, et al. Rheopheresis in clinical practice. Kremlin medicine. Clinical Bulletin. 2004;3:48-53. (In Russ.) Коновалов Г. А., Филоненко И. В., Акопян В. С. и др. Реоферез в клинической практике. Кремлевская медицина. Клинический вестник. 2004;3:48-53.
169. Коновалов Г. А., Чебышев А. Н., Звездкин П. В. и др. Экстракорпоральные методы в лечении тяжелых форм атеросклероза, метаболического синдрома и дилатационной кардиомиопатии. Кремлевская медицина. Клинический вестник. 2001;4:48-54.
д.м.н., профессор, руководитель отдела реабилитации и вторичной профилактики сердечно-сосудистых заболеваний
Москва
д.м.н., профессор, г.н.с., руководитель лаборатории нарушений липидного обмена
Москва
д.м.н., профессор, заслуженный деятель науки РФ, г.н.с.
Москва
д.м.н., профессор, зав. кафедрой кардиологии
Казань
д.м.н., профессор, руководитель отдела атеросклероза, Медицинский институт, ФГБОУ ВО Санкт-Петербургский государственный университет; профессор кафедры госпитальной терапии и кардиологии, ФГБОУ ВО Северо-Западного государственного медицинского университета им. И. И. Мечникова
Санкт-Петербург
д.м.н., профессор, зав. кафедрой пропедевтической терапии с курсом кардиологии, ФГБОУ ВО Самарский государственный медицинский университет Минздрава России; зам. главного врача по медицинской части ГБУЗ Самарский областной клинический кардиологический диспансер им. В. П. Полякова
Самара
д.м.н., профессор кафедры терапии № 1
Краснодар
д.м.н., руководитель лаборатории эпидемиологии питания отдела эпидемиологии хронических неинфекционных заболеваний
Москва
д.м.н., профессор, зав. отделом клинической кардиологии, Научно-исследовательский институт комплексных проблем сердечно-сосудистых заболеваний; профессор кафедры кардиологии и сердечно-сосудистой хирургии
Кемерово
д.м.н., профессор, руководитель Центра диагностики и инновационных медицинских технологий
Москва
д.м.н., руководитель Института персонализированной терапии и профилактики
Москва
д.м.н., профессор, зав. кафедрой госпитальной терапии, Медицинский институт, ФГБОУ ВО Санкт-Петербургский государственный университет Минздрава России; главный врач группы компаний "Мой медицинский центр"
Санкт-Петербург
д.м.н., профессор, профессор кафедры внутренних болезней и семейной медицины
Омск
д.м.н., профессор кафедры кардиологии, г.н.с., руководитель лаборатории фенотипов атеросклероза
Москва
д.м.н., профессор, профессор кафедры госпитальной терапии, Медицинский институт
Санкт-Петербург
Бубнова М.Г., Ежов М.В., Аронов Д.М., Галявич А.С., Гуревич В.С., Дупляков Д.В., Зафираки В.К., Карамнова Н.С., Кашталап В.В., Коновалов Г.А., Мешков А.Н., Обрезан А.Г., Семенкин А.А., Сергиенко И.В., Филиппов А.Е. Гипертриглицеридемия (триглицерид-богатые липопротеины и их ремнанты): роль в развитии атеросклеротических сердечно-сосудистых заболеваний и стратегия контроля. Заключение Комитета экспертов Российского кардиологического общества (РКО), Национального общества по изучению атеросклероза (НОА), Российского общества кардиосоматической реабилитации и вторичной профилактики (РосОКР). Российский кардиологический журнал. 2025;30(5):6364. https://doi.org/10.15829/1560-4071-2025-6364. EDN: VNFGFQ
Bubnova M.G., Ezhov M.N., Aronov D.M., Galyavich A.S., Gurevich V.S., Duplyakov D.V., Zafiraki V.K., Karamnova N.S., Kashtalap V.V., Konovalov G.A., Meshkov A.N., Obrezan A.G., Semenkin A.A., Sergienko I.V., Filippov A.E. Hypertriglyceridemia (triglyceride-rich lipoproteins and their remnants): role in the development of atherosclerotic cardiovascular diseases and control strategy. Opinion of the Expert Committee of the Russian Society of Cardiology, the National Atherosclerosis Society, and the Russian Society of Cardiac and Somatic Rehabilitation and Secondary Prevention. Russian Journal of Cardiology. 2025;30(5):6364. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6364. EDN: VNFGFQ













































