



Содержание
Перейти к:
https://doi.org/10.15829/1560-4071-2025-6179
EDN: EAZOZO
Перейти к:
Многочисленные исследования демонстрируют независимую связь между повышением содержания богатых триглицеридами липопротеидов в крови и риском развития атеросклеротических сердечно-сосудистых заболеваний и острого панкреатита. В обзорной статье детально представлены физиология богатых триглицеридами липопротеинов и патофизиологические аспекты рисков, связанных с гипертриглицеридемиями (ГТГ). Приведены классификации в зависимости от этиологии, фенотипа дислипидемии и степени тяжести. Описаны подходы к диагностике ГТГ.
Семенкин А.А., Мешков А.Н., Ежов М.В. Гипертриглицеридемии — современное состояние вопроса. Часть I: риски, физиология и патофизиологические аспекты, классификация и проблемы диагностики. Российский кардиологический журнал. 2025;30(1):6179. https://doi.org/10.15829/1560-4071-2025-6179. EDN: EAZOZO
Semenkin A.A., Meshkov A.N., Yezhov M.V. Hypertriglyceridemia — current status of the problem. Part I: risks, physiology and pathophysiological aspects, classification and diagnostic problems. Russian Journal of Cardiology. 2025;30(1):6179. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6179. EDN: EAZOZO
Традиционно дислипидемии ассоциируются с холестерином липопротеидов низкой плотности (ЛНП), атеросклерозом и сердечно-сосудистым риском (ССР). В последние годы растет интерес к проблеме гипертриглицеридемий (ГТГ), распростаненность которой в популяции по данным эпидемиологических исследований составляет ~30% [1-4]. Широко обсуждаются ее прогностические аспекты, необходимость и возможности терапии.
Во многих исследованиях продемонстрирована связь между ССР и повышением уровней триглицеридов (ТГ) даже при нормальных значениях холестерина ЛНП и у пациентов с ишемической болезнью сердца, получающих терапию статинами, с достигнутым уровнем холестерина ЛНП 1,0-2,6 ммоль/л [5-8]. Однако по данным большого когортного исследования было показано, что после коррекции по другим факторам риска (возраст, пол, индекс массы тела, потребление алкоголя, сахарный диабет, и др.) небольшое, но значимое повышение риска инфаркта миокарда (в 1,1-1,2 раза) сохраняется только при умеренной и тяжелой (1,7-10,0 ммоль/л) ГТГ и отсутствует при экстремальных значениях ТГ (>10 ммоль/л) [9].
В свою очередь, ГТГ существенно повышает риск развития острого и хронического панкреатита. В структуре причин острого панкреатита ГТГ может занимать до 15% [10]. По данным выше приведенного когортного исследования риск острого панкреатита увеличивается в 1,3, 2,1, 4,3 и 13,6 раза при уровнях ТГ, соответственно, 1,7-4,5 ммоль/л, 4,5-10,0 ммоль/л, 10,0-20,0 ммоль/л и ≥20 ммоль/л, в сравнении с уровнями <1,7 ммоль/л независимо от других факторов [9]. Риск острого панкреатита составляет ~5% и 10-20% при уровнях ТГ >11,3 ммоль/л и >22,6 ммоль/л, соответственно [11]. Таким образом, риск острого панкреатита в отличие от ССР неуклонно увеличивается при любых значениях ТГ, начиная с нормальных, значительно повышается при значениях >10 ммоль/л и становится экстремальным при уровнях ≥20 ммоль/л. Острый панкреатит при ГТГ является потенциально летальным осложнением. Так, в исследовании Gaudet D, et al. смертность, связанная с острым панкреатитом, составила 6,0% при семейной хиломикронемии и 0,55% при многофакторной хиломикронемии [12]. Риск хронического панкреатита, как и острого, значимо повышается при значениях ТГ выше нормальных и возрастает более чем в 6 и 25 раз при уровнях 10-20 ммоль/л и ≥20 ммоль/л, соответственно, в сравнении с лицами, у которых ТГ не превышают 1,7 ммоль/л [9].
На сегодняшний день очевидно, что ГТГ представляют собой неоднородную группу патологий, существенно различающихся по липидным нарушениям и потенциальным рискам, что, несомненно, требует дифференцированных подходов к диагностике и лечению.
ТГ и эфиры холестерина нерастворимы в воде, поэтому циркулируют в кровотоке только в составе липропротеидов, которые представлены амфипатической оболочкой из фосфолипидов с встроенными в нее белками (аполипопротеинами) и свободным холестерином и ядром, содержащим гидрофобные ТГ и эфиры холестерина [13][14].
В определенном количестве ТГ содержатся во всех липопротеидах. Однако основными транспортерами ТГ (богатыми ТГ липопротеидами (БТГЛП)) являются хиломикроны, липопротеиды очень низкой плотности (ЛОНП), продукты их делипидации — ремнанты хиломикронов и ЛОНП и липопротеиды промежуточной плотности (ЛПП) [14-16]. Структурным аполипопротеином БТГЛП и ЛНП является апоB (апоB48 в случае хиломикронов и их ремнантов и апоB100 во всех остальных БТГЛП и ЛНП) [17][18]. Другие аполипопротеины играют существенную роль в их метаболизме и взаимодействии между собой и периферическими тканями [19].
Ключевые моменты метаболизма БТГЛП представлены на рисунке 1. Основной функцией БТГЛП является транспорт ТГ к периферическим тканям. Они имеют двойное происхождение. Экзогенные липиды, поступающие с пищей, транспортируются в составе хиломикронов. Липиды, синтезирующиеся в печени, попадают в кровоток в составе ЛОНП. Сборка хиломикронов происходит в эндоплазматическом ретикулюме энтероцитов на аполипопротеине апоB48 [20]. Наполнение молекулы апоB ТГ катализирует микросомальный белок транспортер ТГ [18][21]. Остальные аполипопротеины хиломикроны получают в процессе сборки (апоAIV и апоAI), либо при взаимодействии с липопротеидами высокой плотности (ЛВП) в плазме крови (апоCII, апоAV, апоCIII, апоCI, апоE, апоAI и апоAII) [18][21][22]. Зрелые хиломикроны попадают в циркуляцию через лимфатическую систему [22]. Сборка ЛОНП в печеночных клетках происходит сходным образом, как и хиломикронов в энтероцитах, с апоB100 и микросомальный белок транспортер ТГ в роли основных участников [23-25]. Остальные аполипопротеины ЛОНП также получают либо в процессе сборки (апоCIII, апоE, апоAV), либо при циркуляции в кровотоке [21][24]. Печеночные клетки могут секретировать два вида ЛОНП: относительно бедные липидами ЛОНП2 и более крупные и богатые ТГ ЛОНП1 [25][26]. Основным стимулятором сборки и секреции хиломикронов и ЛОНП является повышение количества субстрата — свободных жирных кислот, поступающих в процессе пищеварения, при липолизе ТГ в адипоцитах, ремнантах и de novo синтеза в энтероцитах и гепатоцитах [18][27].
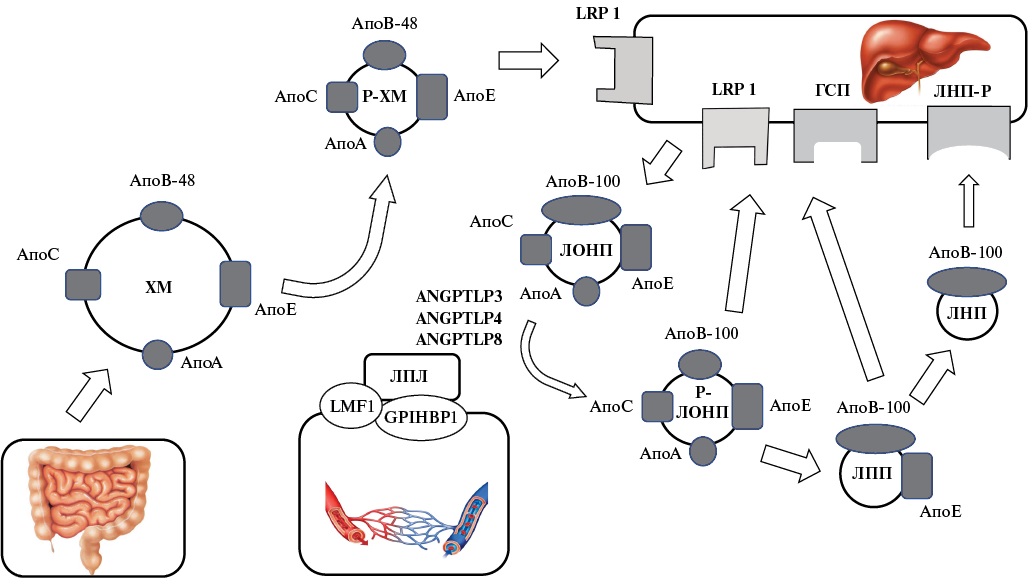
Рис. 1. Ключевые моменты метаболизма БТГЛП.
Сокращения: Апо — аполипопротеины, ГСП — гепарансульфат протеогликаны, ЛНП — липопротеиды низкой плотности, ЛНП-Р — рецептор липопротеидов низкой плотности, ЛОНП — липопротеиды очень низкой плотности, ЛПЛ — липопротеин липаза, ЛПП — липопротеиды промежуточной плотности, Р-ЛОНП — ремнанты липопротеидов очень низкой плотности, Р-ХМ — ремнанты хиломикронов, ХМ — хиломикроны, LRP 1 — белок подобный рецептору ЛНП 1, ANGPTLP — ангиопоэтин подобные протеины, GPIHBP1 — гликозилфосфатидилинозитол-прикрепленный белок, связываюший ЛВП 1, LFM1 — фактор созревания липопротеин липазы 1.
Внутрисосудистый гидролиз ТГ до жирных кислот, содержащихся в хиломикронах и ЛОНП, обусловливающий их делипидацию и конверсию в ремнантные частицы и ЛПП осуществляется липопротеин липазой (ЛПЛ), фиксированной на поверхности эндотелиальных клеток, синтезирующейся в основном адипоцитами, мышечными клетками и макрофагами [28]. АпоB48 и апоB100-содержащие липопротеиды являются конкурентами по взаимодействию с ЛПЛ. В приобретении функциональной структуры ЛПЛ участвует белок — фактор созревания ЛПЛ 1 (LMF1), а гликозилфосфатидилинозитол-прикрепленный белок, связываюший ЛВП 1 (GPIHBP1), отвечает за транслокацию ЛПЛ в просвет капилляра, фиксацию на эндотелии и стабилизацию ее структуры [29]. Снижение активности или дефицит LMF1 и GPIHBP1 вследствие мутаций генов, отвечающих за их синтез, связаны с тяжелой хиломикронемией [30][31]. Имеется ряд регуляторов активности ЛПЛ. АпоCII является основным активатором ЛПЛ, тогда как апоCIII, апоCI, апоE и ангиопоэтин подобные протеины 3, 4 и 8 (ANGPTLP3, ANGPTLP4 и ANGPTLP8) ингибируют ее активность [28][32][33]. АпоAV усиливает липолиз, способствуя прикреплению липопротеидов к эндотелиальным клеткам при взаимодействии с гепарансульфат протеогликанами и GPIHBP1 и подавляет ингибирование ЛПЛ ангиопоэтин подобными протеинами [34-37]. В процессе липолиза ТГ ядро БТГЛП уменьшается на 80-90%, тогда как доля холестерина увеличивается, составляя ~50%, и они превращаются в ремнантные частицы размером <70 нм для хиломикронов и 30-35 нм для ЛОНП [25][38]. Ремнанты хиломикронов элиминируются печенью [24][39]. Ремнантные частицы ЛОНП и ЛПП обладают двумя потенциальными путями метаболизма: дальнейшая делипидация под действием ЛПЛ и печеночной липазы с превращением в ЛНП или прямой клиренс из циркуляции при взаимодействии с рецепторами преимущественно в печени [39-41].
Удаление ремнантов из кровотока осуществляется при взаимодействии лигандов на липопротеидах (апоE, апоB100) со специфическими белками на гепатоцитах (рецептор ЛНП — ЛНП-Р, белок подобный рецептору ЛНП 1 — LRP1, гепарансульфат протеогликаны) [39][42]. Из-за особенностей структуры АпоB48 не способен связываться с ЛНП-Р [43]. Возможность АпоB100 на поверхности ремнантов ЛОНП, являющимся основным лигандом, ответственным за элиминацию ЛНП, взаимодействовать с ЛНП-Р значительно снижена, что связывают с неоптимальной конфигурацией молекулы апоB100 вследствие большего размера частиц ремнантов ЛОНП [44][45]. Таким образом, наличие функционально активных молекул апоE на поверхности ремнантных частиц, которые могут соединяться со всеми тремя видами рецепторов, необходимо для их эффективного клиренса [46]. Взаимодействие ремнантных частиц с гепатоцитами происходит в пространстве Диссе, куда они попадают из печеночных синусоидов через фенестрации в эндотелии [42]. После связывания АпоE со специфическими рецепторами ремнантные частицы подвергаются дальнейшей липолитической трансформации, либо интернализируются гепатоцитами с последующей деградацией в лизосомах [39][42][47].
Важным регулятором обмена липидов является инсулин. В жировых клетках он ингибирует гормон-чувствительную липазу (оказывает антилиполитический эффект), снижая высвобождение свободных жирных кислот в кровеносное русло [48]. В энтероцитах и клетках печени инсулин снижает количество апоB, MТTP и ингибирует продукцию хиломикронов и ЛОНП [18][49][50]. Инсулин ингибирует продукцию апоCIII (ингибитора ЛПЛ) в печени, стимулирует транслокацию печеночного LRP1 на мембрану гепатоцита из внутриклеточных везикул и активирует ЛПЛ в жировой ткани, тем самым ускоряя элиминацию из кровотока БТГЛП [51-53].
Механизмы повышения ССР при ГТГ. На сегодняшний день нет убедительных доказательств, что ТГ сами по себе могут участвовать в процессе атерогенеза [39][54]. Липопротеиды размером <70 нм могут проникать в субэндотелий посредством активного трансцитоза [55]. Соответственно в эту категорию могут попадать все апоB100 содержащие липопротеиды (ЛОНП, ремнанты ЛОНП, ЛПП и ЛНП) и ремнанты хиломикронов, содержащие АпоB48 [39]. Попадая в субэндотелий, БТГЛП могут в неизмененном виде поглощаться макрофагами, запуская процесс образования пенистых клеток — основы формирования атеросклеротической бляшки [56-58]. Другим возможным механизмом ускоренного атерогенеза и дестабилизации покрышки уже имеющейся атеросклеротической бляшки является активизация воспалительного процесса в сосудистой стенке с развитем эндотелиальной дисфункции вследствие высвобождения токсичных свободных жирных кислот при липолизе ТГ БТГЛП, фиксированных в субэндотелии, под действием ЛПЛ макрофагов [39][54].
Считается, что хиломикроны не могут преодолеть эндотелиальный барьер в силу своего размера, потому чистая хиломикронемия (семейная хиломикронемия, I тип дислипидемии по Фридериксону) потенциально не атерогенна [39]. В исследованиях по хиломикронемиям основные сердечно-сосудистые события и наличие ишемической болезни сердца не регистрировались или были минимальными при семейной хиломикронемии в отличие от многофакторной хиломикронемии (V тип дислипидемии по Фридериксону), при которой в крови кроме хиломикронов повышаются другие потенциально атерогенные БТГЛП [59-61].
Дополнительным фактором риска сердечно-сосудистых осложнений при ГТГ является формирование особого липидного профиля — атерогенной дислипидемии, который характеризуется повышением в крови уровня БТГЛП в сочетании с низким уровнем ЛВП и накоплением ЛНП с измененой структурой (мелких плотных ЛНП) [62]. В определенной мере атерогенная дислипидемия является универсальным следствием ГТГ [39]. При высоком уровне в кровотоке БТГЛП под воздействием белка переносчика эфиров холестерина происходит обмен ТГ и холестерина между ЛОНП, ремнантными частицами, ЛНП и ЛВП с увеличением содержания холестерина в ЛОНП и ремнантных частицах и ТГ в ЛНП и ЛВП [63-65]. Обогащенные ТГ ЛНП и ЛВП проходят ускоренную делипидацию в печени под действием печеночной липазы уменьшаясь в размерах [63-65]. Изменение структуры ЛВП нарушает их функцию по обратному транспорту холестерина, способствует их ускоренной элиминации и снижению уровня в кровотоке [66]. Мелкие плотные ЛНП хуже удаляются из кровотока, более подвержены окислению, легче проникают в сосудистую стенку, ускоряя процесс атерогенеза [63][67].
Механизмы повышения риска панкреатита при ГТГ. Точные патофизиологические механизмы, связывающие ГТГ и панкреатит до конца не уточнены. Наиболее распространенная теория развития панкреатита при ГТГ предполагает избыточный гидролиз ТГ БТГЛП до свободных жирных кислот в поджелудочной железе под действием панкреатических липаз, которые в высоких концентрациях могут повреждать панкреатические клетки и капилляры, провоцируя воспалительный процесс [68]. Согласно другой теории, повышение вязкости крови при высоких уровнях крупных БТГЛП (преимущественно хиломикронов) в сыворотке крови приводит к нарушениям микроциркуляции в поджелудочной железе и ишемии с последующим некрозом [68].
С клинической точки зрения актуальны несколько вариантов классификации ГТГ: по уровням ТГ, по спектру липопротеидов крови (по фенотипу), по этиологии.
Для диагностики ГТГ и определения степени ее выраженности, а, соответственно, и потенциальных рисков (сердечно-сосудистая патология или панкреатит) предлагаются классификации, основанные на простом определении уровня ТГ крови натощак. Имеется общий консенсус, что повышенными считаются значения ТГ ≥1,7 ммоль/л, а значения ≥5,7 ммоль/л соответствуют тяжелой ГТГ [39][69]. Европейские эксперты дополнительно предлагают выделять экстремальную ГТГ при значениях >10 ммоль/л [39].
Сохраняет свою актуальность классификация Фридериксона, основанная на составе липопротеидов крови (фенотипе дислипидемии) [70]. Модифицированная классификация, адаптированная к современным реалиям, представлена в таблице 1. В большинстве случаев за исключением IIa типа, при котором повышены только ЛНП, стандартная липидограмма не позволяет определить состав липопротеидов крови и провести точное фенотипирование дислипидемии, что требует проведения электрофореза липопротеидов, либо использования специальных алгоритмов [38, 71]. Так, например, и комбинированная гиперлипидемия (IIb тип) и дисбеталипопротеинемия (III тип) будут проявляться повышением холестерина и ТГ крови, однако состав липопротеидов и патогенез этих нарушений принципиально отличаются. Тем не менее выделение дислипидемий в соответствии с данной классификацией имеет важное практическое значение, т. к. позволяет предположить патогенез имеющихся нарушений, потенциальные риски, обосновать проведение генетических исследований и выбрать оптимальный вариант лечения.
Этиологическая классификация подразумевает деление ГТГ на первичные (генетически опосредованные) и вторичные формы.
Таблица 1
Фенотипы дислипидемий
|
Тип |
Название |
Фенотип |
Повышенные липиды |
|
I |
Хиломикронемия |
ХМ |
ТГ |
|
IIa |
Гиперхолестеринемия (гипербеталипопротеинемия) |
ЛНП |
ХС |
|
IIb |
Комбинированная гиперлипидемия (гипербета- и пребеталипопротеинемия) |
ЛНП, ЛОНП |
ХС и ТГ |
|
III |
Дисбеталипопротеинемия (флотирующие беталипопротеины) |
ремнанты, ЛПП |
ТГ и ХС |
|
IV |
Гипертриглицеридемия (гиперпребеталипопротеинемия) |
ЛОНП |
ТГ и ХС+/- |
|
V |
Многофакторная хиломикронемия (хиломикронемия и гиперпребеталипопротеинемия) |
ХМ, ЛОНП |
ТГ и ХС+/- |
Сокращения: ЛНП — липопротеиды низкой плотности, ЛОНП — липопротеиды очень низкой плотности, ЛПП — липопротеиды промежуточной плотности, ТГ — триглицериды, ХМ — хиломикроны, ХС — холестерин.
Диагностика ГТГ. Для диагностики ГТГ как таковой достаточно рутинного определения ТГ в крови. Одним из диагностических аспектов при ГТГ является правильное определение холестерина ЛНП, особенно при значительно повышенных уровнях ТГ. Имеются непрямые расчетные методы определения холестерина ЛНП на основании определения общего холестерина, холестерина ЛВП и ТГ, использующие различные формулы: Фридвальда, Мартина-Хопкинса и Сэмпсона, которые могут быть использованы при уровнях ТГ <4,5 ммоль/л (для первых двух) и <9,0 ммоль/л (для формулы Сэмпсона) [72-74]. При более высоких значениях ТГ должен использоваться прямой метод определения холестерина ЛНП.
Холестерин не-ЛВП, апоB. Учитывая роль других (помимо ЛНП) апоB100 содержащих липопротеидов в процессе атерогенеза и развитии сердечно-сосудистых осложнений, при наличии ГТГ для определения ССР, резидуального риска на фоне гиполипидемической терапии и выбора тактики лечения необходимо ориентироваться не только на общий холестерин и холестерин ЛНП, но и на холестерин не-ЛВП, определяющийся вычитанием холестерина ЛВП из общего холестерина, отражающий содержание холестерина во всех потенциально атерогенных липопротеидах, или на показатель апоB [75][76]. Принимая во внимание, что апоB-содержащие липопротеиды имеют в составе только одну молекулу этого белка, и на пике их концентрации в постпрандиальном периоде количество апoB100 содержащих липопротеидов, а соответственно, и молекул апoB100, в 9-10 раз превышает количество апoB48 содержащих частиц, и даже при семейной хиломикронемии отношение апoB/апoB48 составляет 7/1, суммарный показатель апoB в большей мере соответствует количеству частиц потенциально атерогенных липопротеидов отличных от хиломикронов [76-78]. Имеются данные, проспективного наблюдения 302430 участников, согласно которым холестерин не-ЛВП и апоB практически идентично предсказывают риск будущих сердечно-сосудистых событий [79]. Более того, потенциально атерогенные (многофакторная хиломикронемия, семейная ГТГ) и атерогенные (дисбеталипопротеинемия) ГТГ обычно не сопровождаются значительным повышением апоB (<120 мг/дл), и использование этого показателя в данных случаях может привести к недооценке риска [38]. В связи с этим рутинная оценка суммарного апоB для определения ССР при наличии ГТГ, особенно при экстремальных значениях ТГ, учитывая дополнительные расходы, представляется менее целесообразной, чем расчет холестерина не-ЛВП.
Диагностика фенотипов ГТГ. Методом диагностики фенотипов ГТГ в клинической практике является электрофорез липопротеидов цельной плазмы крови, который позволяет получить следующую последовательность полосок: хиломикроны; β-полоску, обычно представленную только ЛНП; пре-β-полоску, соответствующую ЛОНП и ремнантным частицам (при дисбеталипопротеинемии) и α-полоску, соответствующую ЛВП. Электрофорез в полиакриламидном геле помимо возможности оценки стандартного спектра липопротеидов позволяет выявлять наличие мелких плотных ЛНП и Лп(а) и проводить количественную оценку [80].
Наличие хиломикронов при выраженной ГТГ подтверждается появлением поверхностного сливкообразного слоя или пленки после выдерживания плазмы крови в холодильнике в течение 18-24 ч при температуре 0-4 ºC [70].
Имеются алгоритмы скрининговой диагностики фенотипов ГТГ, использующие показатели холестерина не-ЛВП и апоB. Так, Sampson M, et al. (2021) предлагают калькулятор (https://figshare.com/articles/software/Sampson_Phenotype_Calculator/16617490), основанный на определении холестерина не-ЛВП [81]. Недостатком метода является невозможность определения дисбеталипопротеинемии (III тип), что подразумевает необходимость оценки клинической симптоматики при использовании данного калькулятора, либо определения дополнительного параметра — отношения холестерин не-ЛВП/апоB, при значениях которого >4,91 дисбеталипопротеинемия подтверждается с чувствительностью 96,8% и специфичностью 95,0% [82]. Скрининговый алгоритм Sniderman A, et al., использующий показатель апоB, позволяет предположить не только фенотип дислипидемии, но и ее этиологию [38]. Алгоритм можно найти по ссылке https://apob.app в виде удобной программы.
Балльная система дифференциальной диагностики экстремальных ГТГ (I и V тип), предполагающая применение лабораторных и клинических данных, предложенная Moulin P, et al., представлена в таблице 2 [83]. При ее использовании диагноз семейной хиломикронемии подтверждается при 10 баллах и выше с чувствительностью 88% и специфичностью 85%, приводимыми авторами, и 96% и 75%, соответственно, по результатам валидации на британской кагорте пациентов с генетически подтвержденным диагнозом семейная хиломикронемия или многофакторная хиломикронемия [83][84].
Таблица 2
Балльная система для прогнозирования вероятности семейной хиломикронемии
|
Баллы |
||
|
1 |
Триглицериды натощак >10 ммоль/л в трех пробах крови, взятых с интервалом не менее 1 мес. Триглицериды натощак >20 ммоль/л как минимум в одной пробе |
+5 +1 |
|
2 |
Уровень триглицеридов, определявшийся ранее, <2 ммоль/л |
-5 |
|
3 |
Отсутствуют вторичные факторыа (за исключением беременностиb и этинилэстрадиола) |
+2 |
|
4 |
Панкреатит в анамнезе |
+1 |
|
5 |
Необъяснимые рецидивирующие абдоминальные боли |
+1 |
|
6 |
В семейном анамнезе отсутствует семейная комбинированная гиперлипидемия |
+1 |
|
7 |
Отсутствует ответ (снижение триглицеридов >20%) на гиполипидемическую терапию |
+1 |
|
8 |
Возраст манифестации симптомов: а) <40 лет б) <20 лет в) <10 лет |
+1 +2 +3 |
Примечание: а — вторичные факторы включают алкоголь, диабет, метаболический синдром, гипотиреоз, кортикостероиды и другие препараты; b — если диагноз ставится во время беременности, необходима повторная оценка после родов. Диагноз ставится на основании суммы баллов: высокая вероятность — ≥10 баллов, маловероятна — ≤9 баллов, очень маловероятна — ≤8 баллов.
Гиперхолестеринемии без ГТГ представляют собой относительно однородную группу патологий, ССР которых связаны с высокими уровнями холестерина ЛНП. ГТГ являются следствием избытка в крови различных БТГЛП, приводящих к вариабельному повышеннию ССР или риска панкреатита, что требует внедрения углубленных диагностических методов для более точного определения характера липидных нарушений. Понимание физиологии метаболизма БТГЛП и патофизиологических аспектов ГТГ позволит врачу взвешенно подходить к выбору тактики обследования пациентов и определению наиболее оптимальных вариантов терапии.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Carroll M, Kit B, Lacher D. Trends in elevated triglyceride in adults: United States, 2001-2012. NCHS Data Brief. 2015;(198):198.
2. Karpov Y, Khomitskaya Y. PROMETHEUS: an observational, cross-sectional, retrospective study of hypertriglyceridemia in Russia. Cardiovasc Diabetol. 2015;14:115. doi:10.1186/s12933-015-0268-2.
3. Мешков А. Н., Ершова А. И., Деев А. И. и др. Распределение показателей липидного спектра у мужчин и женщин трудоспособного возраста в Российской Федерации: результаты исследования ЭССЕ-РФ за 2012-2014гг. Кардиоваскулярная терапия и профилактика. 2017;16(4):62-7. doi:10.15829/1728-8800-2017-4-62-67.
4. Драпкина О. М., Имаева А. Э., Куценко В. А. и др. Дислипидемии в Российской Федерации: популяционные данные, ассоциации с факторами риска. Кардиоваскулярная терапия и профилактика. 2023;22(8S):3791. doi:10.15829/1728-8800-2023-3791.
5. Jeppesen J, Hein HO, Suadicani P, Gyntelberg F. Triglyceride Concentration and Ischemic Heart Disease An Eight-Year Follow-up in the Copenhagen Male Study. Circulation. 1998;97(11):1029-36. doi:10.1161/01.cir.97.11.1029.
6. Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115(4):450-8. doi:10.1161/CIRCULATIONAHA.106.637793.
7. Nichols GA, Philip S, Reynolds K, et al. Increased Cardiovascular Risk in Hypertriglyceridemic Patients With Statin-Controlled LDL Cholesterol. J Clin Endocrinol Metab. 2018;103(8):3019-27. doi:10.1210/jc.2018-00470.
8. Raposeiras-Roubin S, Rossello X, Oliva B, et al. Triglycerides and Residual Atherosclerotic Risk. J Am Coll Cardiol. 2021;77(24):3031-41. doi:10.1016/j.jacc.2021.04.059.
9. Patel RS, Pasea L, Soran H, et al. Elevated plasma triglyceride concentration and risk of adverse clinical outcomes in 1.5 million people: a CALIBER linked electronic health record study. Cardiovasc Diabetol. 2022;21(1):102. doi:10.1186/s12933-022-01525-5.
10. Zhu Y, Pan X, Zeng H, et al. A Study on the Etiology, Severity, and Mortality of 3260 Patients With Acute Pancreatitis According to the Revised Atlanta Classification in Jiangxi, China Over an 8-Year Period. Pancreas. 2017;46(4):504-9. doi:10.1097/MPA.0000000000000776.
11. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195-203. doi:10.1097/01.mcg.0000436438.60145.5a.
12. Gaudet D, Blom D, Bruckert E, et al. Acute Pancreatitis is Highly Prevalent and Complications can be Fatal in Patients with Familial Chylomicronemia: Results from a Survey of Lipidologist. J Clin Lipidol. 2016;10:680-1. doi:10.1016/j.jacl.2016.03.048.
13. Ginsberg HN. Lipoprotein physiology. Endocrinol Metab Clin North Am. 1998;27(3): 503-19. doi:10.1016/s0889-8529(05)70023-2.
14. Северин С. Е. Биологическая химия с упражнениями и задачами. Под ред. С. Е. Северина. Москва: ГЭОТАР-Медиа, 2014. 624 с. ISBN: 978-5-9704-3027-9.
15. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020; 41(1):111-88. doi:10.1093/eurheartj/ehz455.
16. Feingold KR. Lipid and Lipoprotein Metabolism. Endocrinol Metab Clin North Am. 2022;51(3):437-58. doi:10.1016/j.ecl.2022.02.008.
17. Blasiole DA, Davis RA, Attie AD. The physiological and molecular regulation of lipoprotein assembly and secretion. Mol Biosyst. 2007;3(9):608-19. doi:10.1039/b700706j.
18. Dash S, Xiao C, Morgantini C, Lewis GF. New Insights into the Regulation of Chylomicron Production. Annu Rev Nutr. 2015;35:265-94. doi:10.1146/annurev-nutr-071714-034338.
19. Dominiczak MH, Caslake MJ. Apolipoproteins: metabolic role and clinical biochemistry applications. Ann Clin Biochem. 2011;48(Pt 6):498-515. doi:10.1258/acb.2011.011111.
20. Anant S, Davidson NO. Molecular mechanisms of apolipoprotein B mRNA editing. Curr Opin Lipidol. 2001;12(2):159-65. doi:10.1097/00041433-200104000-00009.
21. Gugliucci A. The chylomicron saga: time to focus on postprandial metabolism. Front Endocrinol (Lausanne). 2024;14:1322869. doi:10.3389/fendo.2023.1322869.
22. Pan X, Hussain MM. Gut triglyceride production. Biochim Biophys Acta. 2012;1821(5): 727-35. doi:10.1016/j.bbalip.2011.09.013.
23. Gibbons GF, Wiggins D, Brown AM, Hebbachi AM. Synthesis and function of hepatic very-low-density lipoprotein. Biochem Soc Trans. 2004;32(Pt 1):59-64. doi:10.1042/bst0320059.
24. Ramasamy I. Recent advances in physiological lipoprotein metabolism. Clin Chem Lab Med. 2014;52(12):1695-727. doi:10.1515/cclm-2013-0358.
25. Boren J, Taskinen MR, Packard CJ. Biosynthesis and Metabolism of ApoB-Containing Lipoproteins. Annu Rev Nutr. 2024;44(1):179-204. doi:10.1146/annurev-nutr-062222-020716.
26. Chen J, Fang Z, Luo Q, et al. Unlocking the mysteries of VLDL: exploring its production, intracellular trafficking, and metabolism as therapeutic targets. Lipids Health Dis. 2024;23(1):14. doi:10.1186/s12944-023-01993-y.
27. Tiwari S, Siddiqi SA. Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol. 2012;32(5):1079-86. doi:10.1161/ATVBAHA.111.241471.
28. Wu SA, Kersten S, Qi L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol Metab. 2021;32(1):48-61. doi:10.1016/j.tem.2020.11.005.
29. Young SG, Fong LG, Beigneux AP, et al. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019;30(1):51-65. doi:10.1016/j.cmet.2019.05.023.
30. Peterfy M, Ben-Zeev O, Mao HZ, et al. Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet. 2007;39(12):1483-7. doi:10.1038/ng.2007.24.
31. Beigneux AP, Davies BS, Gin P, et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5(4):279-91. doi:10.1016/j.cmet.2007.02.002.
32. Wolska A, Reimund M, Remaley AT. Apolipoprotein C-II: the re-emergence of a forgotten factor. Curr Opin Lipidol. 2020;31(3):147-53. doi:10.1097/MOL.0000000000000680.
33. Mehta A, Shapiro MD. Apolipoproteins in vascular biology and atherosclerotic disease. Nat Rev Cardiol. 2022;19(3):168-79. doi:10.1038/s41569-021-00613-5.
34. Shu X, Nelbach L, Weinstein MM, et al. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav-/-mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler Thromb Vasc Biol. 2010;30(12):2504-9. doi:10.1161/ATVBAHA.110.210815.
35. Merkel M, Loeffler B, Kluger M, et al. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280(22):21553-60. doi:10.1074/jbc.M411412200.
36. Li Y, He PP, Zhang DW, et al. Lipoprotein lipase: from gene to atherosclerosis. Atherosclerosis. 2014;237(2):597-608. doi:10.1016/j.atherosclerosis.2014.10.016.
37. Yang Y, Konrad RJ, Ploug M, Young SG. APOA5 deficiency causes hypertriglyceridemia by reducing amounts of lipoprotein lipase in capillaries. J Lipid Res. 2024;65(7):100578. doi:10.1016/j.jlr.2024.100578.
38. Sniderman A, Couture P, de Graaf J. Diagnosis and treatment of apolipoprotein B dyslipoproteinemias. Nat Rev Endocrinol. 2010;6(6):335-46. doi:10.1038/nrendo.2010.50.
39. Ginsberg HN, Packard CJ, Chapman MJ, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42(47):4791-806. doi:10.1093/eurheartj/ehab551.
40. Santamarina-Fojo S, González-Navarro H, Freeman L, et al. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24(10):1750-4. doi:10.1161/01.ATV.0000140818.00570.2d.
41. Packard CJ, Boren J, Taskinen MR. Causes and Consequences of Hypertriglyceridemia. Front Endocrinol (Lausanne). 2020;11:252. doi:10.3389/fendo.2020.00252.
42. Williams KJ, Chen K. Recent insights into factors affecting remnant lipoprotein uptake. Curr Opin Lipidol. 2010;21(3):218-28. doi:10.1097/MOL.0b013e328338cabc.
43. Veniant MM, Zlot CH, Walzem RL, et al. Lipoprotein clearance mechanisms in LDL receptor-deficient "Apo-B48-only" and "Apo-B100-only" mice. J Clin Invest. 1998;102(8):1559-68. doi:10.1172/JCI4164.
44. Bradley WA, Hwang SL, Karlin JB, et al. Low-density lipoprotein receptor binding determinants switch from apolipoprotein E to apolipoprotein B during conversion of hypertriglyceridemic very-low-density lipoprotein to low-density lipoproteins. J Biol Chem. 1984;10;259(23):14728-35.
45. Koopal C, Marais AD, Westerink J, Visseren FL. Autosomal dominant familial dysbetalipoproteinemia: A pathophysiological framework and practical approach to diagnosis and therapy. J Clin Lipidol. 2017;11(1):12-23.e1. doi:10.1016/j.jacl.2016.10.001.
46. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. 2019;51(2):165-76. doi:10.1016/j.pathol.2018.11.002.
47. Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507-37. doi:10.1146/annurev.genom.1.1.507.
48. Recazens E, Mouisel E, Langin D. Hormone-sensitive lipase: sixty years later. Prog Lipid Res. 2021;82:101084. doi:10.1016/j.plipres.2020.101084.
49. Malmstrom R, Packard CJ, Caslake M, et al. Effects of insulin and acipimox on VLDL1 and VLDL2 apolipoprotein B production in normal subjects. Diabetes. 1998;47(5):779-87. doi:10.2337/diabetes.47.5.779.
50. Verges B. Abnormal hepatic apolipoprotein B metabolism in type 2 diabetes. Atherosclerosis. 2010;211(2):353-60. doi:10.1016/j.atherosclerosis.2010.01.028.
51. Robinson DS, Speake BK. Role of insulin and other hormones in the control of lipoprotein lipase activity. Biochem Soc Trans. 1989;17(1):40-2. doi:10.1042/bst0170040.
52. Chen M, Breslow JL, Li W, Leff T. Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: correlation with changes in plasma triglyceride levels. J Lipid Res. 1994;35(11):1918-24.
53. Laatsch A, Merkel M, Talmud PJ, et al. Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis. 2009;204(1):105-11. doi:10.1016/j.atherosclerosis.2008.07.046.
54. Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res. 2016;118(4): 547-63. doi:10.1161/CIRCRESAHA.115.306249.
55. Boren J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-30. doi:10.1093/eurheartj/ehz962.
56. Brown ML, Ramprasad MP, Umeda PK, et al. A macrophage receptor for apolipoprotein B48: cloning, expression, and atherosclerosis. Proc Natl Acad Sci U S A. 2000;97(13): 7488-93. doi:10.1073/pnas.120184097.
57. Takahashi S, Sakai J, Fujino T, et al. The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor. J Atheroscler Thromb. 2004;11(4):200-8. doi:10.5551/jat.11.200.
58. Llorente-Cortes V, Badimon L. LDL receptor-related protein and the vascular wall: implications for atherothrombosis. Arterioscler Thromb Vasc Biol. 2005;25(3):497-504. doi:10.1161/01.ATV.0000154280.62072.fd.
59. Paquette M, Bernard S, Hegele RA, Baass A. Chylomicronemia: Differences between familial chylomicronemia syndrome and multifactorial chylomicronemia. Atherosclerosis. 2019;283:137-42. doi:10.1016/j.atherosclerosis.2018.12.019.
60. O'Dea LSL, MacDougall J, Alexander VJ, et al. Differentiating Familial Chylomicronemia Syndrome From Multifactorial Severe Hypertriglyceridemia by Clinical Profiles. J Endocr Soc. 2019;3(12):2397-410. doi:10.1210/js.2019-00214.
61. Belhassen M, Van Ganse E, Nolin M, et al. 10-Year Comparative Follow-up of Familial versus Multifactorial Chylomicronemia Syndromes. J Clin Endocrinol Metab. 2021; 106(3):e1332-e1342. doi:10.1210/clinem/dgaa838.
62. Stahel P, Xiao C, Hegele RA, Lewis GF. The Atherogenic Dyslipidemia Complex and Novel Approaches to Cardiovascular Disease Prevention in Diabetes. Can J Cardiol. 2018;34(5):595-604. doi:10.1016/j.cjca.2017.12.007.
63. Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43(9):1363-79. doi:10.1194/jlr.r200004-jlr200.
64. Rashid S, Watanabe T, Sakaue T, Lewis GF. Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin Biochem. 2003;36(6):421-9. doi:10.1016/s0009-9120(03)00078-x.
65. Hirano T. Pathophysiology of Diabetic Dyslipidemia. J Atheroscler Thromb. 2018;25(9): 771-82. doi:10.5551/jat.RV17023.
66. Srivastava RAK. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol Cell Biochem. 2018;440(1-2):167-87. doi:10.1007/s11010-017-3165-z.
67. Diffenderfer MR, Schaefer EJ. The composition and metabolism of large and small LDL. Curr Opin Lipidol. 2014;25(3):221-6. doi:10.1097/MOL.0000000000000067.
68. Kiss L, Fűr G, Pisipati S, et al. Mechanisms linking hypertriglyceridemia to acute pancreatitis. Acta Physiol (Oxf). 2023;237(3):e13916. doi:10.1111/apha.13916.
69. Virani SS, Morris PB, Agarwala A, et al. 2021 ACC Expert Consensus Decision Pathway on the Management of ASCVD Risk Reduction in Patients With Persistent Hypertriglyceridemia: A Report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2021;78(9):960-93. doi:10.1016/j.jacc.2021.06.011.
70. Beaumont JL, Carlson LA, Cooper GR, et al. Classification of hyperlipidaemias and hyperlipoproteinaemias. Bull World Health Organ. 1970;43(6):891-915.
71. Koopal C, Marais AD, Visseren FL. Familial dysbetalipoproteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes. 2017;24(2):133-9. doi:10.1097/MED.0000000000000316.
72. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499-502.
73. Martin SS, Blaha MJ, Elshazly MB, et al. Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA. 2013;310(19):2061-8. doi:10.1001/jama.2013.280532.
74. Sampson M, Ling C, Sun Q, et al. A New Equation for Calculation of Low-Density Lipoprotein Cholesterol in Patients With Normolipidemia and/or Hypertriglyceridemia. JAMA Cardiol. 2020;5(5):540-8. doi:10.1001/jamacardio.2020.0013.
75. Raja V, Aguiar C, Alsayed N, et al. Non-HDL-cholesterol in dyslipidemia: Review of the state-of-the-art literature and outlook. Atherosclerosis. 2023;383:117312. doi:10.1016/j.atherosclerosis.2023.117312.
76. Glavinovic T, Thanassoulis G, de Graaf J, et al. Physiological Bases for the Superiority of Apolipoprotein B Over Low-Density Lipoprotein Cholesterol and Non-High-Density Lipoprotein Cholesterol as a Marker of Cardiovascular Risk. J Am Heart Assoc. 2022;11(20):e025858. doi:10.1161/JAHA.122.025858.
77. Blom DJ, O'Dea L, Digenio A, et al. Characterizing familial chylomicronemia syndrome: Baseline data of the APPROACH study. J Clin Lipidol. 2018;12(5):1234-1243.e5. doi:10.1016/j.jacl.2018.05.013.
78. Elovson J, Chatterton JE, Bell GT, et al. Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. J Lipid Res. 1988;29(11):1461-73.
79. Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993-2000. doi:10.1001/jama.2009.1619.
80. Blom DJ, Byrnes P, Jones S, Marais AD. Non-denaturing polyacrylamide gradient gel electrophoresis for the diagnosis of dysbetalipoproteinemia. J Lipid Res. 2003;44(1): 212-7. doi:10.1194/jlr.d200013-jlr200.
81. Sampson M, Ballout RA, Soffer D, et al. A new phenotypic classification system for dyslipidemias based on the standard lipid panel. Lipids Health Dis. 2021;20(1):170. doi:10.1186/s12944-021-01585-8.
82. Boot CS, Middling E, Allen J, Neely RDG. Evaluation of the non-HDL cholesterol to apolipoprotein B ration as a screening test for dysbetalipoproteinemia. Clin Chem. 2019;65(2):313-20. doi:10.1373/clinchem.2018.292425.
83. Moulin P, Dufour R, Averna M, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an "FCS score". Atherosclerosis. 2018;275:265-72. doi:10.1016/j.atherosclerosis.2018.06.814.
84. Bashir B, Kwok S, Wierzbicki AS, et al. Validation of the familial chylomicronaemia syndrome (FCS) score in an ethnically diverse cohort from UK FCS registry: Implications for diagnosis and differentiation from multifactorial chylomicronaemia syndrome (MCS). Atherosclerosis. 2024;391:117476. doi:10.1016/j.atherosclerosis.2024.117476.
Д.м.н., профессор, профессор кафедры внутренних болезней и семейной медицины ДПО.
Омск
Нет
Д.м.н., руководитель Института персонализированной терапии и профилактики.
Москва
Нет
Д.м.н., г.н.с., руководитель лаборатории нарушений липидного обмена Научно-исследовательского института клинической кардиологии им. А.Л. Мясникова.
Москва
Нет
Семенкин А.А., Мешков А.Н., Ежов М.В. Гипертриглицеридемии — современное состояние вопроса. Часть I: риски, физиология и патофизиологические аспекты, классификация и проблемы диагностики. Российский кардиологический журнал. 2025;30(1):6179. https://doi.org/10.15829/1560-4071-2025-6179. EDN: EAZOZO
Semenkin A.A., Meshkov A.N., Yezhov M.V. Hypertriglyceridemia — current status of the problem. Part I: risks, physiology and pathophysiological aspects, classification and diagnostic problems. Russian Journal of Cardiology. 2025;30(1):6179. (In Russ.) https://doi.org/10.15829/1560-4071-2025-6179. EDN: EAZOZO













































